ADP-ribosylation is a reversible post-translational protein modification, which is evolutionarily conserved in prokaryotic and eukaryotic organisms. It governs critical cellular functions, including, but not limited to cellular proliferation, differentiation, RNA translation, and genomic repair. The addition of one or multiple ADP-ribose moieties can be catalysed by poly(ADP-ribose) polymerase (PARP) enzymes, while in eukaryotic organisms, ADP-ribosylation can be reversed through the action of specific enzymes capable of ADP-ribose signalling regulation. In several lower eukaryotic organisms, including Trypanosomatidae parasites, ADP-ribosylation is thought to be important for infection establishment. Trypanosomatidae encompasses several human disease-causing pathogens, including Trypanosoma cruzi, T. brucei, and the Leishmania genus. These parasites are the etiological agents of Chagas disease, African trypanosomiasis (sleeping sickness), and leishmaniasis, respectively.
- ADP-ribosylation
- PARP
- PARG
- Trypanosoma
- Leishmania
1. ADP-Ribosylation in Infection
1.1. ADP-Ribosylation in Viral Pathogens
1.2. ADP-Ribosylation in Bacterial Pathogens
Studies examining the roles of PARPs during bacterial pathogen infection are not as extensive in comparison to those on PARPs in viral infections. Only a small number of bacterial species possess functional PARylation systems. Although the majority contain domains capable of PAR binding, in addition to PAR-degrading enzymes [18]. The majority of antibacterial studies on PARP enzymes have primarily focused on the use of PARP1. Studies have been performed on several notable human bacterial pathogens, including Helicobacter, Salmonella, Escherichia coli, Pseudomonas aeruginosa, Streptococcus pneumonia, Streptococcus pyogenes, and Chlamydophila. Experiments utilising these bacteria surmise a similar consensus, whereby shifts in PARP activity increase the difficulty of mounting an effective response in preventing damage from bacterial infection [19][20][21][22][23][19,20,21,22,23]. Several species of bacteria have also been found to possess PARG enzymes, including Thermomonospora curvata and Herpetosiphon aurantiacus [24][27]. In humans, PARG enzymes are a mechanism through which PAR can be removed from the cell via catabolism of poly(ADP-ribose), through hydrolysis of the ribose-ribose bonds. This prevents damage caused by excessive PAR accumulation in the cytoplasm [25][28]. PAR accumulation can lead to a PARP-mediated cell death pathway known as parthanatos, through which excessive PAR can lead to apoptosis via several mechanisms, including depletion of NAD and the PAR-mediated activation of an apoptosis-inducing factor (AIF) [26][29]. PAR can bind to AIF, which is followed by AIF translocation to the nucleus, resulting in extensive DNA fragmentation and chromatin damage [27][30]. PARG is the primary means through which excessive PAR is removed in human cells. Other human PAR hydrolases do exist, necessitated by PARG’s inability to remove the most proximal ADP-ribose moieties [28][31], including ARH3, which is present during the removal of PAR from the mitochondria [29][32]. There is much evidence to suggest the extensive roles of human PARP enzymes in immune protection during bacterial and viral infection. However, key questions remain for both. PARP activity is seemingly broader across enzymes in terms of antiviral activity (10 out of 17 human PARP enzymes have identified antiviral activity). Potentially as a result of primarily cytoplasmic and nuclear localisation, which allows the PARP enzymes to interrupt viral replication cycles at several distinct stages [30][34]. Therefore, it seems that the expression of multiple cytoplasmic PARP enzymes, developed alongside the evolution of vertebrates, are seemingly as equally as important as the nuclear-localised PARPs in maintaining cellular health through the maintenance of essential processes and antimicrobial activity.2. Trypanosomatidae
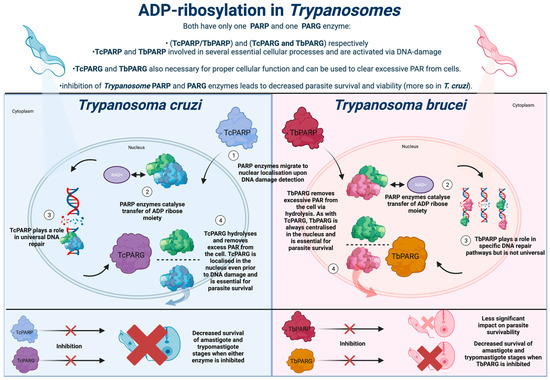
Trypanosomatidae is an order of singular flagellate kinetoplastid parasites, the most relevant to human health being Trypanosoma and Leishmania. The parasites T. cruzi, T. brucei, and Leishmania are all responsible for infections categorised by the World Health Organisation (WHO) as neglected tropical diseases (NTDs) [31][37]. As such, these infections are responsible for profound impacts as they primarily arise in vulnerable people inhabiting developing countries. ADP-ribosylation and the associated enzymes, including PARP and PARG, play an essential role in the ability of Trypanosomatidae to establish a successful infection in the human host. Given the lack of safe and effective medications available to combat these parasites, and the parasites’ reliance on the functioning ADP-ribosylation to maintain parasitic viability, the disruption of their ADP-ribosylation systems may present a novel and effective therapeutic option. T. cruzi is the causative agent of Chagas disease, which is endemic to Central and South America. An initial infection during the acute stage results in flu-like systems. These symptoms mask the diagnosis of T. cruzi infection [32][38]. During chronic stages, an infection can lay dormant for decades, characterised by potentially minimal symptoms and low parasitemia, which causes issues in detecting the infection. The chronic infection eventually results in organ enlargement, with parasites primarily targeting the heart, leading to numerous cardiac complications and potential death. T. brucei is the etiological agent of African trypanosomiasis. Vectoral transmission is the most common route of infection, in which parasites enter the human host via bites inflicted by the primary tsetse fly host. Similar to Chagas disease, the initial stage of infection presents aspecific symptoms, followed by parasites eventually migrating to the brain, leading to neurological complications, and ultimately death, without proper treatment [33][41]. Available therapies for the neurological stage of infection are also limited, with Melarsoprol the only available drug, although this causes death in 5% of the patients who ingest it since Melarsoprol resistance is present in some strains [34][42]. The anti-parasitic Fexinidazole has shown activity against both the CNS and peripheral stages of African trypanosomiasis, although studies remain in clinical trials and effective drug alternatives are required to limit resistance [35][43]. Leishmaniasis is an umbrella term for three distinct diseases: visceral, cutaneous, and mucocutaneous leishmaniasis. Infections are predominantly found in Asia, the Middle East, and Northern Africa to differing degrees, The diseases are caused by several different Leishmania species and are transmitted commonly by bite wounds inflicted by an infected female phlebotomine sand fly. Visceral leishmaniasis is the most severe, as it causes a systemic infection that almost invariably is fatal without rapid treatment. The cutaneous infection leads to superficial skin lesions, whilst mucocutaneous infection leads to significant damage of the buccal and nasal cavities via the formation of damaging mucocutaneous lesions, which may disappear and reoccur repeatedly [36][44]. Given the severe nature of the infection, the majority of attention in the development of novel therapies for leishmaniasis has been focused on visceral leishmaniasis [37][38][45,46]. Consequently, given the scarcity of funding available to research the development of novel therapeutics to combat these NTDs and the lack of efficacy for existing treatments, the identification of novel ways to combat these infections is paramount. T. cruzi, T. brucei, and Leishmania all utilise ADP-ribosylation in several distinct ways to facilitate successful infection in the human host. Given the avenues with potential to be explored, whereby ADP-ribosylation manipulation can be harnessed to combat viral and bacterial infection, it is feasible that inhibiting or disrupting ADP-ribosylation in these parasites could lead to diminished parasite survival and proliferation. In contrast to the extensive PARP network found in humans, the PARP system present in Trypanosomatidae is much simpler. T. cruzi and T. brucei both possess a singular PARP enzyme, designated TcPARP and TbPARP, respectively. Both parasites also utilise a sole poly(ADP-ribose) glycohydrolase (PARG) enzyme, which is used to reverse the action of PARP via the hydrolysis of ribose–ribose bonds present in PARP, which helps prevent extensive DNA damage by excessive PARP accumulation [39][49]. The primary ADP-ribosylation mechanisms within these parasites use polyADP-ribosylation, although there is also evidence of them using monoADP-ribosylation systems. Both T. cruzi and T. brucei possess proteins that are homologous to human MacroD1 and MacroD2, which are domains that hydrolyse and cleave ADP-ribose attachments to proteins in monoADP-ribosylation within human systems [40][50].3. ADP-Ribosylation in Trypanosoma cruzi
3.1. Use of ADP-Ribosylation and Associated PARP and PARG Enzymes in Trypanosoma cruzi
Trypanosoma cruzi expresses a sole PARP enzyme throughout its lifecycle, known as TcPARP (Figure 1). An initial study of TcPARP revealed several structural and molecular similarities to its human homologue, hPARP-1 [41][52]. Similar to hPARP-1, TcPARP is activated via DNA strand nicks, upon exposure of the parasite DNA to damaging agents such as H2O2. Initial studies on TcPARP revealed a highly evolutionarily conserved C-terminal catalytic domain that is homologous to T. brucei PARP, human PARP-1, and PARP-4. Furthermore, the catalytic triad of histidine, tyrosine, and glutamic acid is utilised for PAR elongation in hPARP-1, the closest human homologue to TcPARP is conserved within the parasite [42][53].