Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Argen Mamazhakypov and Version 2 by Alfred Zheng.
Pulmonary hypertension (PH) is a pathological condition with multifactorial etiology, which is characterized by elevated pulmonary arterial pressure and pulmonary vascular remodeling. Accumulating clinical evidence suggests that circulating osteopontin may serve as a biomarker of PH progression, severity, and prognosis, as well as an indicator of maladaptive right ventricular remodeling and dysfunction. Osteopontin modulates a plethora of cellular processes within the pulmonary vasculature, including cell proliferation, migration, apoptosis, extracellular matrix synthesis, and inflammation via binding to various receptors such as integrins and CD44.
- osteopontin
- pulmonary hypertension
- right heart failure
1. Introduction
Pulmonary hypertension (PH) is a condition affecting pulmonary vasculature [1] and is hemodynamically defined as a mean pulmonary artery pressure (mPAP) greater than 20 mmHg at rest as assessed by right heart catheterization [2]. PH is classified into five main clinical groups: pulmonary arterial hypertension (PAH), PH resulting from left heart disease, PH resulting from chronic lung disease or hypoxia, chronic thromboembolic PH (CTEPH), and PH with unclear or multifactorial mechanisms [2]. PH represents a chronic and ultimately fatal pulmonary vascular disorder of multifactorial origin [3]. The primary pathological characteristics of PH include persistent pulmonary vascular constriction and excessive obstructive pulmonary vascular remodeling [4]. At the cellular level, initial pathological mechanisms of pulmonary vascular remodeling are characterized by dysfunction and apoptosis of pulmonary artery endothelial cells (PAECs). At later stages, hyperproliferation and apoptosis resistance of pulmonary artery smooth muscle cells (PASMCs) lead to structural changes in the pulmonary vasculature, elevation in pulmonary arterial pressure (PAP), and pulmonary vascular resistance, which ultimately culminate in right ventricular (RV) failure [5].
2. Osteopontin Signaling
The name “osteopontin”, which is composed of two words, was proposed by Oldberg and colleagues [6][15]. The prefix osteo- is derived from “osteon”, the Greek word for bone, and reflects the fact that osteopontin was first isolated from the mineralized bone matrix of bovines as a bone sialoprotein I [7][16]. The suffix -pontin is derived from “pons”, the Latin word for bridge, and reflects osteopontin’s role as a linking protein between cells and hydroxyapatite in the matrix [6][15]. Osteopontin is also known as secreted phosphoprotein 1, uropontin, and early T-lymphocyte activation-1. As soon as osteopontin was isolated, it was revealed that the matricellular protein osteopontin is a cytokine that is synthesized and expressed by a wide array of cells and tissues in the body. The tissues and organs expressing osteopontin include kidney, inner ear, brain, heart, lung, vessels, skin, and bone marrow [8][17], as well as luminal epithelial surfaces of the gastrointestinal tract, gall bladder, pancreas, urinary and reproductive tracts, lung, breast, salivary glands, and sweat glands [9][18]. Osteopontin is also found in biological fluids, such as blood, milk, urine, and seminal fluid [8][17]. Osteopontin expression is considerably upregulated in response to a variety of pathological processes, including inflammation, mechanical stress, and tissue injury and repair. In various cardiovascular diseases, synthesis of osteopontin is induced in smooth muscle cells and cardiomyocytes [8][17]. Osteopontin plays a multifarious role in biological processes such as inflammation, immunological response, wound healing, cellular adhesion, migration, survival, and biomineralization. Binding of osteopontin to integrins activates signaling pathways that regulate diverse cellular functions including cell proliferation, adhesion, invasion, migration, and fibrosis [10][19]. Osteopontin also interacts with CD44, a ubiquitously expressed cell-surface receptor, which exists as a number of isoforms [11][20]. It is encoded by 20 exons, 10 of which are constant and form the invariant extracellular domain of the smallest standard isoform [12][21]. The variant isoforms are generated by alternative splicing and may contain in addition to 10 constant exons a single variant exon or a combination of variant exons [12][21]. CD44 has been shown to be involved in cell–matrix and cell–cell interactions [13][22]. Although hyaluronic acid is considered as a principal ligand for CD44, other extracellular-matrix proteins including serglycin, collagen, fibronectin, chondroitin sulfate, laminin, and osteopontin also may serve as ligands for CD44 [13][22]. Osteopontin does not interact with the standard isoform of CD44, but rather binds to its variant isoforms [14][23], such as CD44v6 and CD44v7 [15][16][24,25]. Variant CD44 isoforms play critical roles in the development of various cancers through their interaction with osteopontin [12][21]. The exact role of the osteopontin-mediated activation of CD44 variants in cardiovascular diseases remains poorly explored and the literature is scarce. It was demonstrated that the osteopontin–CD44v6 interaction mediates calcium deposition in valve interstitial cells from patients with noncalcified aortic valve sclerosis [17][26]. Histological studies revealed CD44 expression in lung plexiform lesions from patients with idiopathic PAH (IPAH) [18][27]. Another study showed that adventitial fibroblasts isolated from chronically hypoxic hypertensive calves displayed increased expression of CD44 along with αVβ3 and osteopontin [19][28]. However, these studies did not investigate which CD44 isoforms were upregulated, and did not demonstrate an interaction between CD44 and osteopontin. Recently, CD44v7–10, CD44v8–10, CD44v9–10, and CD44v10 transcripts were detected in lungs of mice subjected to chronic hypoxia [20][29]. In PAH patients, the CD44v8–10 variant was expressed by endothelial-to-mesenchymal transition-like PAECs in pulmonary vessels with neointimal hyperplasia or occluded vessels, including plexiform lesions [20][29]. It remains, however, to be elucidated whether osteopontin interacts with these CD44 variants and which signaling pathways are activated in the setting of PH. Recent studies showed that osteopontin serves as a ligand for CD275, which regulates immune responses by activated T cells [21][30]. Alternative splicing of osteopontin generates three isoforms: osteopontin-a (the full-length isoform), osteopontin-b (lacking exon 5), and osteopontin-c (lacking exon 4) [22][31]. Alternative translation of osteopontin produces two isoforms: a cell-secreted full-length and a shortened intracellular protein lacking the N-terminal signal sequence [22][31]. In addition, osteopontin undergoes numerous posttranslational modifications, such as serine/threonine phosphorylation, sulfation, O-glycosylation, glutamination, and proteolytic processing, which can subsequently determine the functional variability of osteopontin [23][32]. To date, thrombin, matrix metalloproteinases (MMPs), caspase-8/3, plasmin, cathepsin D, and enterokinase have been identified as proteases that cleave osteopontin at different sites, resulting in the formation of several fragments of different sizes and with variable functions [24][33]. Taken together, the ultimate roles and functions of osteopontin are impacted by multiple factors, spanning from gene transcription to protein translation, posttranslational modification, and proteasomal processing of the final protein product. Osteopontin-mediated cellular signaling pathways are well explored in cancer diseases. Thus, interaction of osteopontin with cell surface receptors, integrins, and/or CD44 activates JNK, Ras/Raf/MEK/ERK, PI3K/AKT, JAK/STAT, NF-κB, and TIAM1/Rac1 signaling pathways, leading to enhancement of various malignant properties of cancer cells [25][34]. Unfortunately, the role of signaling pathways activated by osteopontin in pulmonary hypertension has been poorly studied.3. Osteopontin in Pulmonary Vascular Cells
3.1. Osteopontin in Pulmonary Artery Endothelial Cells
Osteopontin plays a crucial role in both the physiology and pathophysiology of endothelial cells in the systemic vasculature. It serves as a vital mediator in the intricate physiological and pathological processes that govern endothelial cells in the pathogenesis of several cardiovascular diseases. Various factors, including aldosterone [26][58], vascular endothelial cell growth factor (VEGF) [27][59], and hypoxia [28][60], regulate osteopontin expression in endothelial cells, and vice versa, it regulates various functions of endothelial cells. Osteopontin stimulates endothelial cell differentiation and angiogenesis via its SVVYGLR fragment [29][61]. It promotes angiogenesis by mediating endothelial cell attachment to the ECM [30][62] and synthesis of angiogenic factors by endothelial cells [31][63]. Osteopontin enhances VEGF expression through AKT and ERK signaling pathways to induce endothelial cell motility, proliferation, and tube formation via the αvβ3-integrin receptor [32][64]. In turn, VEGF augments expression of osteopontin and αvβ3-integrin in endothelial cells and stimulates integrin-dependent endothelial cell migration [27][59]. A conserved sequence comprising nine amino acids (RSKSKKFRR) located at the thrombin cleavage site of osteopontin promotes endothelial cell migration, proliferation, and tube formation in vitro [33][65]. The crucial function of osteopontin in neovascularization is further corroborated by the findings of the compromised angiogenic capacity of endothelial cells derived from osteopontin knockout mice, which is partially restored by exogenous administration of osteopontin [34][66]. Besides angiogenesis, osteopontin influences several other endothelial cell functions. It increases vascular permeability by downregulating expression of the tight junction proteins ZO-1 and claudin-5 in endothelial cells [35][67], induces endothelial-to-mesenchymal transition via CD44 receptor in response to shear stress [36][68] and regulates endothelial cell apoptosis [37][69]. The precise role of osteopontin in the functional regulation of PAECs and its interplay in the pathogenesis of pulmonary vascular remodeling still remains largely unknown. It can be hypothesized that elevated circulating osteopontin levels in PH patients may exert effects on PAECs similar to those on endothelial cells in systemic vasculature, potentially leading to alterations in endothelial cell physiology leading to subsequent pathological pulmonary vascular remodeling. This is due to the established fact that endothelial cell dysfunction plays a crucial role in the pathogenesis of PH, particularly in the early stages of the disease [38][70]. However, the precise mechanisms by which osteopontin affects PAEC physiology and its potential contribution to the development of PH remains a subject of future research.3.2. Osteopontin in Pulmonary Artery Smooth Muscle Cells
Osteopontin critically influences physiology of systemic vascular smooth muscle cells (SMCs) through modulation of a myriad of cellular processes, including cell proliferation [39][71], apoptosis [40][72], migration [41][42][73,74], and neointima formation [43][75]. Various factors, including hypoxia [44][76], platelet-derived growth factor (PDGF) [45][77], hyperglycemia [46][78], mechanical stress [47][79], aldosterone [48][80], and reactive oxygen species (ROS) [49][81], regulate osteopontin expression in SMCs. In vitro studies revealed that the cell proliferation rate of SMCs is directly related to osteopontin expression levels in these cells [50][82], suggesting a direct role of osteopontin in mediating cell proliferation. Autocrine expression of osteopontin contributed to PDGF-induced SMC migration [51][83] and regulated adhesion of SMCs to the ECM [52][84] and production of MMPs [53][85]. Osteopontin increased ECM synthesis in SMCs via activating the p38 MAPK signaling pathway [54][86]. Production of osteopontin in SMCs was inhibited by a cyclic guanosine monophosphate (cGMP)-dependent protein kinase, an important mediator of nitric oxide (NO) and cGMP signaling [55][87]. Pharmacological agents enhancing NO-cGMP signaling might thus potentially attenuate osteopontin expression. Available data suggest that osteopontin regulates various cellular functions of PASMCs, similar to the systemic vascular SMCs. Osteopontin was found to be as one of the most highly overregulated matricellular proteins in senescent PASMCs [56][40]. Its expression in these cells was associated with an augmented cell migration and proliferation [56][40], which were markedly reduced in the presence of neutralizing anti-osteopontin antibodies [56][40]. Multiple factors with known pathological roles in the pulmonary vascular remodeling, including acidic fibroblast growth factor [57][88], angiotensin-II [57][88], transforming growth factor-β [58][89], PDGF-BB [59][90], hypoxia [60][61][91,92], sphingosine-1-phosphate [62][93], and mechanical stretch [63][42], induce osteopontin expression in PASMCs through various signaling pathways. Acidic fibroblast growth factor induced osteopontin expression in PASMCs via activating Ras/JNK and Ras/MEK1/2 signaling pathways [57][88], while hypoxia did so via ERK and p38MAPK signaling pathways [61][92] and mechanical stretch via AKT-ERK signaling pathways [63][42]. In turn, osteopontin modulates various functions of PASMCs. In particular, it promoted PASMC proliferation and migration in a dose-dependent manner via αVβ3-integrin mediated AKT and ERK1/2 signaling pathways [63][42]. Furthermore, activation of the calcineurin/NFATc3 signaling pathway by sphingosine-1-phosphate upregulated osteopontin expression and stimulated PASMCs proliferation, which was suppressed by the activation of the proliferator-activated receptor gamma (PPAR-γ) [62][93].3.3. Osteopontin in Pulmonary Artery Adventitial Fibroblasts
Recent research demonstrated the significance of vascular adventitia and adventitial fibroblasts in maintaining vascular homeostasis of both systemic [64][94] and pulmonary vasculature [65][95]. The adventitial fibroblasts are involved in the regulation of various cellular processes, including ECM remodeling, inflammation, and angiogenesis, which are critical for maintaining the structural and functional integrity of the vasculature [64][65][94,95]. In the systemic vasculature, osteopontin plays a significant role in SMC-mediated regulation of adventitial fibroblast functions [66][96]. Integrin β3 was found to be the responsible receptor for promoting osteopontin-mediated adventitial fibroblast migration [67][97]. The role of osteopontin in the physiology of pulmonary vascular adventitial fibroblasts was studied primarily in the bovine model of PH. Adventitial fibroblasts isolated from calves with severe hypoxia-induced PH exhibited high proliferative, migratory, and pro-invasive capabilities. These functional alterations of adventitial fibroblasts were mediated via activated ERK1/2 and AKT signaling pathways that correlated with high osteopontin expression [19][28]. Inhibition of osteopontin expression with a specific small interfering RNA or neutralizing antibodies led to an attenuation of proliferative, migratory, and invasive capabilities of adventitial fibroblasts [19][28].3.4. Osteopontin in Pulmonary Vascular Macrophages
The literature pertaining to the specific functions of osteopontin in the functional dynamics of macrophages and their interactions in the pathogenesis of PH is scarce. In hypoxia, osteopontin can act as an inflammatory cytokine, thereby promoting the chemotaxis of various inflammatory cells and modulating the inflammatory milieu in the affected tissue. Although circulating monocytes do not express osteopontin, it is among the most highly expressed genes in activated macrophages [68][98]. Additionally, it serves as a powerful chemotactic stimulus for these immune cells [69][99]. Osteopontin regulates key functions of macrophages, including migration, survival, phagocytosis, and pro-inflammatory cytokine production [70][71][100,101]. These effects are mainly mediated via interaction with α4/α9-integrins by its SLAYGLR domain [72][102]. The potent pro-inflammatory properties of osteopontin are further augmented following its cleavage by thrombin, which leads to the generation of an N-terminal fragment containing two integrin-binding domains, namely the RGD and SVVYGLR motifs [73][103]. Osteopontin can contribute to pulmonary vascular remodeling through facilitating attraction and retention of macrophages and T lymphocytes in areas of inflammation within pulmonary vessels [74][104]. A single-cell RNA sequencing demonstrated that enhanced osteopontin expression in lung tissues from patients with systemic sclerosis was enriched in macrophages [68][98], suggesting that osteopontin signaling in these cells might contribute to the pulmonary vascular remodeling in PAH associated with systemic sclerosis. Thus, available data suggest that osteopontin might contribute to the chemotaxis of leukocytes and other inflammatory cells to the remodeled pulmonary vasculature and thereby be involved in the pathogenesis of the disease. However, the intricacies of osteopontin function within pulmonary vascular macrophages and other immunocompetent cells and their contribution to pulmonary vascular remodeling remain enigmatic, warranting future investigation.3.5. Osteopontin in Intercellular Communications of Vascular Cells in Pulmonary Vascular Remodeling
Elevated circulating osteopontin levels in PH can be attributed to its enhanced synthesis by pulmonary vascular cells. The intricate regulation of osteopontin expression by various factors including hypoxia, growth factors, mechanical stress, and inflammation in these cells highlights its complexity. Direct effects of osteopontin on pulmonary vascular cells through both paracrine and autocrine mechanisms suggest its potential role in regulating cellular proliferation, migration, and other functions in these cells, thereby potentially serving as a key mediator in intercellular communication within the pulmonary vasculature (Figure 1). These effects of osteopontin on pulmonary vascular cells adversely affect the disease and may determine the impact of circulating osteopontin on the status and outcome of PAH patients. However, the intricacy of these interactions, particularly concerning the extent of their contribution and hierarchical involvement in the pathological processes of pulmonary vascular remodeling, presents a significant challenge in precisely defining the cell-specific role of osteopontin in pulmonary vascular cells.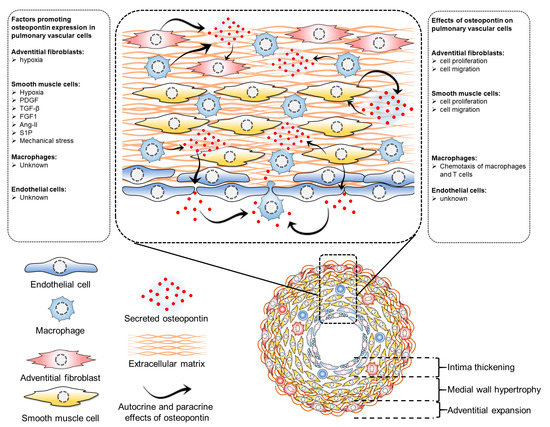
Figure 1. Regulation of osteopontin expression and effects of osteopontin in pulmonary vascular cells. Several factors regulate osteopontin expression in pulmonary vascular cells. In adventitial fibroblasts, hypoxia induces osteopontin expression, which has implications in cell proliferation and migration. In pulmonary artery smooth muscle cells, many factors regulate osteopontin expression, including hypoxia, PDGF (platelet-derived growth factor), TGF-beta (transforming growth factor beta), FGF1 (fibroblast growth factor 1), Ang-II (angiotensin II), S1P (sphingosine 1-phosphate), and mechanical stress, all of which also regulate cell proliferation and migration. However, the exact factors regulating osteopontin in pulmonary vascular endothelial cells and macrophages have not been identified. While osteopontin regulates macrophage chemotaxis, its role in endothelial cells remains unknown.
4. Osteopontin in Right Ventricular Remodeling
Numerous investigations utilizing both in vivo animal models and human clinical studies have demonstrated a crucial role for osteopontin in the pathogenesis of LV failure of various etiologies [75][112]. However, there remains a paucity of data pertaining to the specific role of osteopontin in RV dysfunction and failure. Accumulating evidence suggests that osteopontin may play a significant role in RV pathologies. Similar to the pulmonary vascular remodeling, remodeled RV may represent an important source of circulating osteopontin. A correlation between the plasma concentrations of osteopontin and the expression levels within the hypertrophied RV myocardium was demonstrated in monocrotaline and SuHx rat PH models [76][77][113,114]. Recent transcriptomic analysis of RV tissues from monocrotaline and SuHx rats revealed that osteopontin was one of the top nine overregulated genes [78][115], suggesting that pressure overload induces osteopontin expression in the RV.
The proposition that osteopontin may negatively impact the RV is partially predicated upon the findings of various studies that reported a reduction in osteopontin expression following treatment with pharmacological agents that improve pulmonary hemodynamics and RV function, such as PPAR-γ activator pioglitazone [79][109] and estrogen receptor-β agonist 17β-estradiol [76][113]. While it is possible that this decrease in osteopontin expression is simply a reflection of the reduced afterload, which subsequently mitigates the stress exerted upon the RV wall, it cannot be entirely ruled out that these agents may exert a direct impact on the RV and thereby attenuate osteopontin expression. Therefore, it is crucial to perform further investigations utilizing the afterload-independent experimental models of RV failure, in which the concentrations of osteopontin are either enhanced via exogenous administration or decreased through the utilization of genetically modified knockout organisms or neutralizing antibodies, in order to either confirm or refute such hypotheses.
5. Osteopontin as a Treatment Target in Pulmonary Hypertension
Despite the wealth of data implicating osteopontin in the pathogenesis of PH, studies employing specific therapeutic approaches directly targeting osteopontin, such as osteopontin-neutralizing antibodies or osteopontin aptamers in preclinical PH models, are still lacking. Studies based on the cancer pathologies demonstrated a therapeutic potential of various approaches to suppress osteopontin using aptamers, antibodies, and small molecular as well as the miRNA-based medications [80][116]. Among these many options, aptamers have emerged as one of the promising strategies to target osteopontin more specifically and precisely. Osteopontin aptamers effectively block osteopontin function in vitro [81][117] and in vivo [82][118] and thereby provide therapeutic benefits. Similarly, osteopontin-neutralizing antibodies suppressed osteopontin in various heart failure models [83][84][119,120]. Blocking osteopontin receptors represents another interesting approach to inhibit its effects. Application of an αVβ3-integrin antagonist accelerated recovery from the pulmonary vascular remodeling and RV failure in surgically corrected shunt rats [63][42]. Taken together, despite the accumulating evidence implicating osteopontin in the pathogenesis of PH, the field has yet to fully explore the potential therapeutic utility of osteopontin inhibition through various modalities, including but not limited to: RNA interference utilizing small interfering RNAs, short hairpin RNAs, aptamers, monoclonal antibodies, and small molecular inhibitors. Further investigations in animal models are imperative in order to evaluate the potential benefits of osteopontin inhibition in the management of PH.