Metformin is a metabolic modulator widely used to threat type II diabetes and metabolic syndrome patients. It shows a safe profile and iIts use in additional indications, such as cancer, is an important matter of investigation.
- Metabolism, Diabetes, Oncology
1. Introduction
Metformin (N′,N′-dimethylbiguanide) represents the most frequently administered drug to treat patients with metabolic syndrome and type 2 diabetes, worldwide. Its use has spanned over 60 years and is partially due to a very positive risk–benefit profile [1][2][3][4]. Systemically, metformin therapy lowers blood glucose in type 2 diabetes patients by targeting hepatic gluconeogenesis and by increasing glucose uptake in the peripheral tissues, mainly muscles, and indirectly reduces the insulin blood levels by counteracting insulin resistance[3][5]. Before digging into the relationships between metformin and cancer, we believe it may be useful to quickly review the main mechanism of action (MoA) of metformin in diabetic, non-cancer patients.
Undisputedly, the finding that metformin indirectly activates the adenosine 5′-monophosphate protein kinase (AMPK)[6][7][8], represented an important turn in the still unfinished quest for its mechanism of action (MoA)[9] (Figure 1). The “secret recipe” in metformin’s MoA consists of its hydrophilic nature, cationic behavior, Fe and Cu-binding properties and a pKa within the physiological pH range[10]. Metformin accumulates in the mitochondria of intact cells by virtue of its positive charge[11](Figure 1), causing inhibition of complex 1 of the respiratory chain [12]. In detail, metformin interferes with the coupling of redox and proton transfer domains in complex 1, resulting in altered redox status at the mitochondria and cytosol [10][13] and reactive oxygen species (ROS) accumulation [11][14]. The reduced phosphorylation of adenine nucleotides and accumulation of AMP allosterically determines the liver kinase B1-STE20-related pseudokinase-calcium binding protein-39 (LKB1-STRAD-CAB39)-mediated activation of AMPK[15]. Furthermore, “non-canonical” activation of AMPK takes place at lysosomes and is triggered either by changes in fructose-1,6-bisphosphate (F1, 6P2) which affects LKB1-mediated phosphorylation [16], or by galectin-9-promoted transforming growth factor-β-activated kinase-1 (TAK1)-mediated phosphorylation [17], the latter being linked to induction of autophagy[18][19]. Interestingly, activation of AMPK seems to be spatially and temporally regulated: a mild increase in AMP may activate the cytoplasmic and lysosomal pool while a more sustained increase in AMP may promote phosphorylation of AMPK by the LKB1 complex in mitochondria[20]. This may have a functional consequence in light of the topographic constraints and gradients existing within both normal tissues and in the tumor mass.
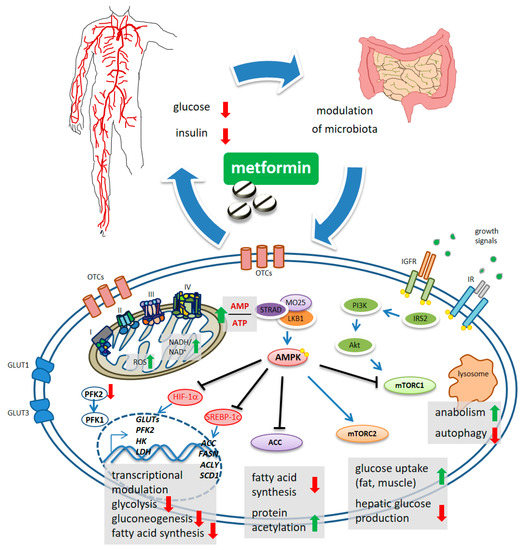
Figure 1. Schematic model of metformin action. Systemically, metformin lowers the glucose levels and, indirectly, reduces insulin levels. Metformin crosses the plasma membrane at least partially through organic cation transporters (OCTs and related) and enters mitochondria where it affects complex 1 coupling and causes altered redox status and increased AMP/ATP ratio. This latter activates, allosterically, the kinase LKB1 which phosphorylates AMPK. mTORC1 functions as an environmental sensor and is activated by insulin signaling and growth factors to promote anabolic metabolism and to inhibit autophagy. Metformin-stimulated AMPK reverses mTORC1 actions. mTORC2 is activated by AMPK and stimulates increased glucose entry into muscles and reduced glucose production in the liver. ACC inhibition reduces fatty acid synthesis and may increase collective protein acetylation (increased acetyl-CoA) thus exerting transcriptional modulation. Metformin-stimulated-AMPK controls transcriptionally hepatic gluconeogenesis. Metformin interferes with the increase in HIF-1α and the control of glycolytic genes and GLUT transporters, including the expression of PFK2 which increases the levels of the metabolite fructose-2,6-P2, allosterically activating PFK-1. Downregulated expression of ACC, FASN, ACLY and SCD1 was promoted by metformin, through interference with SREBP-1c and TR4.
Regarding the inhibition of gluconeogenesis, recent evidence shows that the early, acute downregulation of gluconeogenesis is AMPK independent and is possibly linked to compromised functioning of complex 1 that causes unbalanced NADH/NAD in mitochondria and an altered cytosolic redox state[20][21] and/or by inhibition of the mitochondrial glycerol-phosphate dehydrogenase (mGPD)[22].
The late inhibition of gluconeogenesis happens transcriptionally and is AMPK dependent. In this latter process, the small heterodimer partner (SHP), a transcriptional co-repressor, and phosphorylation of CREB binding protein (CBP) by AMPK-PKCι/λ (protein kinase C) were shown to play critical roles[23][24][25][26] (Figure 1). Additionally, even with some context specificity, AMPK targets additional metabolic enzymes such as HK2, glycogen synthase (GS), and hydroxy-methyl-glutaryl-CoA reductase (HMGCR)[27][28]. Metformin also downregulated the expression of glucose transporters (GLUT1, GLUT3) and of glycolytic enzymes such as hexokinase 2 (HK2), 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 (PFKFB4), pyruvate kinase (PKM) and lactate dehydrogenase A(LDH)[29] . In hepatocarcinoma cells (HCC), metformin inhibited phosphofructokinase-1 (PFK1) activity by suppressing the expression of PFK2, thereby reducing the allosteric activation of PFK1 by fructose-2,6-bisphosphate[30] (Figure 1). These effects were achieved through metformin-induced inhibition of HIF-1α activity and its binding to the hypoxia-responsive elements (HRE) within the promoter region of these genes[31]. Collectively, the effect of metformin-activated AMPK accounts for increased catabolism and decreased anabolism by modulating protein synthesis, lipid homeostasis, glycolysis—and mitochondrial homeostasis, in addition to transcriptionally modulating gluconeogenesis (Figure 1).
2. Metformin Modulates the Activity of mTORC1 and mTORC2 Complexes
Mammalian target of rapamycin (mTOR) is the core of two, functionally distinct, multiprotein complexes, mTOR complex 1 (mTORC1) and mTORC2[32], oppositely modulated by metformin via AMPK[33][34]. mTORC1 activation exerts anabolic effects (through increased ribosome biogenesis, lipid, nucleotide and protein synthesis) and suppresses autophagy. The mTORC1 is activated by insulin and growth factors via phosphatidyl-inositol 3-kinase (PI3K)/AKT[34]. Activated AMPK directly phosphorylates tuberous sclerosis complex 2 (TSC2), thereby inhibiting mTORC1 [[33]. mTORC1 signaling can also be inhibited by a metformin-sensitive Ras-related GTPase, as shown in mouse embryo fibroblasts[35]. On the other hand, metformin-stimulated AMPK activates mTORC2, thereby promoting cell survival and systemically reducing hepatic glucose production[36].
3. Metformin Inhibits Fatty Acid Synthesis
Another direct AMPK target protein highly relevant for cellular energy consumption, is the acetyl-CoA carboxylase (ACC), deactivated by AMPK via phosphorylation[37]. The inhibition of ACC activity decreased fatty acid synthesis consequent to a reduced conversion of acetyl-CoA to malonyl-CoA[38] (Figure 1). ACC inhibition increases collective protein acetylation and thus, may exert transcriptional modulation[37][38] (Figure 1). Additionally, phosphorylation by metformin-activated AMPK inhibited the proteasome-mediated degradation of insulin-induced gene 1 (Insig-1), which in turn reduced the activating cleavage of the transcription factor sterol regulatory element-binding protein-1c (SREBP-1c) and consequently, reduced lipogenic gene expression[3938] (Figure 1). In a mouse hepatoma model, metformin decreased de novo fatty acid synthesis by reducing, transcriptionally, the expression of acetyl CoA carboxylase, fatty acid synthase (FASN) and ATP citrate lyase (ACLY)[40]. Additionally, metformin impaired—AMPK-dependently—the binding and transactivation of the nuclear receptor TR4 to its responsive elements in the stearoyl-CoA desaturase-1 (SCD1) promoter, in hepatocytes[41] (Figure 1).
4. Metformin Modulates Gut Microbiota
[39]
Remodeling the gut microbiota mediates the therapeutic effects of metformin and is responsible for its known gastrointestinal side effect[43]s[42][43]. In fact, there is long-known evidence showing that the full glucose-lowering effect of metformin is bound to its oral administration [44] and that antibiotics may blunt the effect of metformin in animal models[45]. Additionally, the concentration of metformin in the jejunum was estimated to be from ten to a few hundred times higher than in plasma[46]. The changes in microbiota elicited by metformin in diabetic patients can be relevant for its anticancer ac[47]tion, given the involvement of gut microbiota in the pathogenesis of colorectal cancer (CRC) and other solid tumors[47]. For instance, an effect of metformin on the abundance of intestinal Akkermansia muciniphila has been reproducibly reported[45][[48]. A. Muciniphila was shown to increase the abundance of gut-targeted CD4+ T cells, providing an adjuvant effect to the action of anti- programmed cell death -1 (PD-1) agents, in animal models of melanoma and non-small cell lung cancer (NSCLC)[49] and in a model of microsatellite-stable (MSS) colorectal cancer[50].
References
- Alusik, S.; Paluch, Z. Metformin: The past, presence, and future. Minerva Med. 2015, 106, 233–238.
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576.
- Rojas, L.B.; Gomes, M.B. Metformin: An old but still the best treatment for type 2 diabetes. Diabetol. Metab. Syndr. 2013, 5, 6.
- Sterne, J. Treatment of diabetes mellitus with N,N-dimethylguanylguanidine (LA. 6023, glucophage). Therapie 1959, 14, 625–630.
- Agius, L.; Ford, B.E.; Chachra, S.S. The Metformin Mechanism on Gluconeogenesis and AMPK Activation: The Metabolite Perspective. Int. J. Mol. Sci. 2020, 21, 3240.
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174.
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646.
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585.
- Spiering, M.J. The mystery of metformin. J. Biol. Chem. 2019, 294, 6689–6691.
- Cameron, A.R.; Logie, L.; Patel, K.; Erhardt, S.; Bacon, S.; Middleton, P.; Harthill, J.; Forteath, C.; Coats, J.T.; Kerr, C.; et al. Metformin selectively targets redox control of complex I energy transduction. Redox Biol. 2018, 14, 187–197.
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487.
- Andrzejewski, S.; Gravel, S.P.; Pollak, M.; St-Pierre, J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014, 2, 12.
- Logie, L.; Harthill, J.; Patel, K.; Bacon, S.; Hamilton, D.L.; Macrae, K.; McDougall, G.; Wang, H.H.; Xue, L.; Jiang, H.; et al. Cellular responses to the metal-binding properties of metformin. Diabetes 2012, 61, 1423–1433.
- Bridges, H.R.; Sirvio, V.A.; Agip, A.N.; Hirst, J. Molecular features of biguanides required for targeting of mitochondrial respiratory complex I and activation of AMP-kinase. BMC. Biol. 2016, 14, 65.
- Hardie, D.G. Keeping the home fires burning: AMP-activated protein kinase. J. R. Soc. Interface 2018, 15.
- Zhang, C.S.; Li, M.; Zong, Y.; Lin, S.C. Determining AMPK Activation via the Lysosomal v-ATPase-Ragulator-AXIN/LKB1 Axis. Methods Mol. Biol. 2018, 1732, 393–411.
- Jia, J.; Bissa, B.; Brecht, L.; Allers, L.; Choi, S.W.; Gu, Y.; Zbinden, M.; Burge, M.R.; Timmins, G.; Hallows, K.; et al. AMPK is activated during lysosomal damage via a galectin-ubiquitin signal transduction system. Autophagy 2020, 16, 1550–1552.
- Carroll, B.; Dunlop, E.A. The lysosome: A crucial hub for AMPK and mTORC1 signalling. Biochem. J. 2017, 474, 1453–1466.
- Jia, J.; Bissa, B.; Brecht, L.; Allers, L.; Choi, S.W.; Gu, Y.; Zbinden, M.; Burge, M.R.; Timmins, G.; Hallows, K.; et al. AMPK, a Regulator of Metabolism and Autophagy, Is Activated by Lysosomal Damage via a Novel Galectin-Directed Ubiquitin Signal Transduction System. Mol. Cell 2020, 77, 951–969 e959.
- Zong, Y.; Zhang, C.S.; Li, M.; Wang, W.; Wang, Z.; Hawley, S.A.; Ma, T.; Feng, J.W.; Tian, X.; Qi, Q.; et al. Hierarchical activation of compartmentalized pools of AMPK depends on severity of nutrient or energy stress. Cell Res. 2019, 29, 460–473.
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.M.; Zhang, D.; Camporez, J.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat. Med. 2018, 24, 1384–1394.
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546.
- He, L.; Sabet, A.; Djedjos, S.; Miller, R.; Sun, X.; Hussain, M.A.; Radovick, S.; Wondisford, F.E. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 2009, 137, 635–646.
- Rines, A.K.; Sharabi, K.; Tavares, C.D.; Puigserver, P. Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nat. Rev. Drug Discov. 2016, 15, 786–804.
- Kim, Y.D.; Park, K.G.; Lee, Y.S.; Park, Y.Y.; Kim, D.K.; Nedumaran, B.; Jang, W.G.; Cho, W.J.; Ha, J.; Lee, I.K.; et al. Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes 2008, 57, 306–314.
- Lee, J.M.; Seo, W.Y.; Song, K.H.; Chanda, D.; Kim, Y.D.; Kim, D.K.; Lee, M.W.; Ryu, D.; Kim, Y.H.; Noh, J.R.; et al. AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB.CRTC2 complex by orphan nuclear receptor small heterodimer partner. J. Biol. Chem. 2010, 285, 32182–32191.
- Hardie, D.G. AMP-activated protein kinase: Maintaining energy homeostasis at the cellular and whole-body levels. Annu. Rev. Nutr. 2014, 34, 31–55.
- Ruderman, N.B.; Cacicedo, J.M.; Itani, S.; Yagihashi, N.; Saha, A.K.; Ye, J.M.; Chen, K.; Zou, M.; Carling, D.; Boden, G.; et al. Malonyl-CoA and AMP-activated protein kinase (AMPK): Possible links between insulin resistance in muscle and early endothelial cell damage in diabetes. Biochem. Soc. Trans. 2003, 31, 202–206.
- Tyszka-Czochara, M.; Bukowska-Strakova, K.; Kocemba-Pilarczyk, K.A.; Majka, M. Caffeic Acid Targets AMPK Signaling and Regulates Tricarboxylic Acid Cycle Anaplerosis while Metformin Downregulates HIF-1alpha-Induced Glycolytic Enzymes in Human Cervical Squamous Cell Carcinoma Lines. Nutrients 2018, 10, 841.
- Hu, L.; Zeng, Z.; Xia, Q.; Liu, Z.; Feng, X.; Chen, J.; Huang, M.; Chen, L.; Fang, Z.; Liu, Q.; et al. Metformin attenuates hepatoma cell proliferation by decreasing glycolytic flux through the HIF-1alpha/PFKFB3/PFK1 pathway. Life Sci. 2019, 239, 116966.
- Semenza, G.L. Transcriptional regulation by hypoxia-inducible factor 1 molecular mechanisms of oxygen homeostasis. Trends Cardiovasc. Med. 1996, 6, 151–157.
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825.
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab. 2017, 25, 463–471.
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal. 2019, 12.
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brule, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010, 11, 390–401.
- Fu, W.; Hall, M.N. Regulation of mTORC2 Signaling. Genes (Basel) 2020, 11, 1045.
- Angin, Y.; Beauloye, C.; Horman, S.; Bertrand, L. Regulation of Carbohydrate Metabolism, Lipid Metabolism, and Protein Metabolism by AMPK. Exp. Suppl. 2016, 107, 23–43.
- Abu-Elheiga, L.; Matzuk, M.M.; Abo-Hashema, K.A.; Wakil, S.J. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 2001, 291, 2613–2616.
- Han, Y.; Hu, Z.; Cui, A.; Liu, Z.; Ma, F.; Xue, Y.; Liu, Y.; Zhang, F.; Zhao, Z.; Yu, Y.; et al. Post-translational regulation of lipogenesis via AMPK-dependent phosphorylation of insulin-induced gene. Nat. Commun. 2019, 10, 623.
- Bhalla, K.; Hwang, B.J.; Dewi, R.E.; Twaddel, W.; Goloubeva, O.G.; Wong, K.K.; Saxena, N.K.; Biswal, S.; Girnun, G.D. Metformin prevents liver tumorigenesis by inhibiting pathways driving hepatic lipogenesis. Cancer Prev. Res. (Phila) 2012, 5, 544–552.
- Kim, E.; Liu, N.C.; Yu, I.C.; Lin, H.Y.; Lee, Y.F.; Sparks, J.D.; Chen, L.M.; Chang, C. Metformin inhibits nuclear receptor TR4-mediated hepatic stearoyl-CoA desaturase 1 gene expression with altered insulin sensitivity. Diabetes 2011, 60, 1493–1503.
- Bryrup, T.; Thomsen, C.W.; Kern, T.; Allin, K.H.; Brandslund, I.; Jorgensen, N.R.; Vestergaard, H.; Hansen, T.; Hansen, T.H.; Pedersen, O.; et al. Metformin-induced changes of the gut microbiota in healthy young men: Results of a non-blinded, one-armed intervention study. Diabetologia 2019, 62, 1024–1035.
- Wu, T.; Horowitz, M.; Rayner, C.K. New insights into the anti-diabetic actions of metformin: From the liver to the gut. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 157–166.
- Sum, C.F.; Webster, J.M.; Johnson, A.B.; Catalano, C.; Cooper, B.G.; Taylor, R. The effect of intravenous metformin on glucose metabolism during hyperglycaemia in type 2 diabetes. Diabet Med. 1992, 9, 61–65.
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735.
- Bailey, C.J.; Wilcock, C.; Scarpello, J.H. Metformin and the intestine. Diabetologia 2008, 51, 1552–1553.
- Cheng, W.Y.; Wu, C.Y.; Yu, J. The role of gut microbiota in cancer treatment: Friend or foe? Gut 2020, 69, 1867–1876.
- de la Cuesta-Zuluaga, J.; Mueller, N.T.; Corrales-Agudelo, V.; Velasquez-Mejia, E.P.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Metformin Is Associated With Higher Relative Abundance of Mucin-Degrading Akkermansia muciniphila and Several Short-Chain Fatty Acid-Producing Microbiota in the Gut. Diabetes Care 2017, 40, 54–62.
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97.
- Xu, X.; Lv, J.; Guo, F.; Li, J.; Jia, Y.; Jiang, D.; Wang, N.; Zhang, C.; Kong, L.; Liu, Y.; et al. Gut Microbiome Influences the Efficacy of PD-1 Antibody Immunotherapy on MSS-Type Colorectal Cancer via Metabolic Pathway. Front. Microbiol 2020, 11, 814.