Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Alessandra Pulliero and Version 2 by Conner Chen.
Ferroptosis is an iron-dependent form of programmed cell death. Defined by a novel term for the first time by Dixon et al. in 2012, it has been studied before and has been described in the last decade as a different type of cell death with respect to apoptosis, necrosis, and autophagy, from which it differs from genetic, biochemical, and morphological point of views.
- ferroptosis
- microRNA
- human cancer
1. Ferroptotic Pathway
Ferroptosis is an iron-dependent form of programmed cell death. Defined by a novel term for the first time by Dixon et al. in 2012 [1][6], it has been studied before [2][3][4][7,8,9] and has been described in the last decade as a different type of cell death with respect to apoptosis, necrosis, and autophagy, from which it differs from genetic, biochemical, and morphological point of views.
The term “ferroptosis” was coined following the observations that RAS-mutated cancer cells were more sensitive to ferroptosis activation compared to cancer cells that lack RAS mutations [1][5][6,10]. This could be explained by the tight connection between oncogenic RAS activation and the high levels of intracellular iron in these cells, since the oncogenic RAS modulates iron metabolism through the regulation of transferrin receptor 1 (TFR1) [4][6][7][9,11,12]. However, it has been observed that, while mutated RAS could be necessary to initiate the ferroptosis mechanism, several kinds of cancer are also sensitive to ferroptosis induction, even if they lack RAS mutations [8][13]. In contrast, two important inducers of ferroptosis such as Erastin and RSL3 (oncogenic-RAS-selective lethal compounds) are known to specifically induce this kind of cell death in RAS-mutant cancer cells following the accumulation of reactive oxygen species by an iron-dependent mechanism [9][10][14,15].
Recent studies reported that the well-known tumor suppressor gene p53 plays a role in ferroptosis [11][16]. P53 was first reported to sensitize cells to ferroptosis through transcriptionally repressing SLC7A11 which representsone of its direct targets [12][17]. The regulation of ferroptisis by p53 contributes to the tumor suppressive function of p53 itself. Notably, tumors expressing mutant p53 may display decreased levels of SLC7A11 and increased sensitivity to ferroptosis [13][18]. The role of p53 in ferroptosis is rather complex implying both a promotion as well as a suppression of the process, likely in a cell-context-dependent manner and involving different finely tuned proteins and pathways with regulation that is not discussed in this review.
Ferroptosis is the consequence of an imbalance between the levels of reactive oxygen species (ROS) and the activity of the antioxidant defense, with a consequent accumulation of ROS in the plasmatic membrane, process widely known for causing many disorders in the cell [14][19]. In this context, a significant intermediate role is played by the different components of iron metabolism. Therefore, there are three mechanisms that, when deregulated, have a key role in triggering the ferroptosis machinery: iron metabolism, the antioxidant defense, and lipid metabolism (Figure 1).
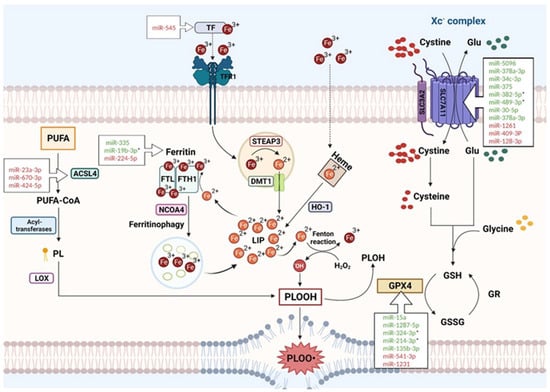
Figure 1. Interplay of the three main pathways contributing to ferroptosis and principal miRNAs targeting key ferroptotic proteins (see text for description). * miRNA reported as having a therapeutic impact. Created with https://www.BioRender.com (accessed on 20 April 2023).
2. Iron Metabolism
Ferroptosis is an iron-dependent type of cell death; therefore, the intracellular levels of iron play a major role in its induction. The crucial mediators are a group of factors that regulate iron at different levels inside and outside the cell. In the blood, iron circulates in the Fe3+ form carried by a special transport protein called transferrin (TF). When it must enter the cell, Fe3+ is carried by transferrin near the plasmatic membrane, where the transferrin receptor 1 (TFR1) recognizes the complex and allows the Fe3+ to enter by endocytosis. Inside the endosome, Fe3+ is reduced to Fe2+ by the metal reductase Six-Transmembrane Epithelial Antigen of Prostate 3 (STEAP3) [15][20]. Fe2+ can then be released in the cytoplasm by the Divalent Metal Transporter (DMT1) and enter the labile iron pool (LIP) [16][21]. When the Fe2+ in the LIP reaches high levels, it can be stored mainly in ferritin or as heme.
Ferritin is an iron storage protein complex formed by two subunits consisting of a ferritin light chain (FTL) and a ferritin heavy chain 1 (FTH1) [17][18][22,23]. The FTH1 subunit has a ferroxidase activity that is able to oxidase the ferrous iron into the ferric form in which it can be stored [19][20][24,25]. The mediator that regulates the release of Fe2+ from the ferritin storage is the nuclear receptor co-activator 4 (NCOA4) that is able to bind the heavy chain of the ferritin complex and promote the ferritin degradation mediated by the lysosomes (“ferritinophagy”) [21][22][26,27]. Any overactivity of the NCOA4 can cause a major ferritin degradation, leading to an increase in the intracellular LIP. In contrast, some type of cells, such as macrophages, enterocytes, red blood cells, and neurons, export iron on the outside; only one iron exporter is known, called ferroportin [23][28].
The regulation of intracellular iron levels is controlled by iron-responsive element-binding proteins IRP1 and IRP2 [24][29]: when the iron decreases, IRP1 and IRP2 can bind the iron-responsive elements (IREs) located on the UTR regions of the mRNA [25][30]. Ferritin and ferroportin contain the IREs in the UTR regions, so when the cell is in a state of iron deficiency, IRPs repress their synthesis [25][30].
The other iron storage protein, heme, can release the Fe2+ when induced by heme oxygenase-1 (HO-1) [26][27][31,32]. The increase in Fe2+ in the cellular LIP can be one of the first events triggering the ferroptosis machinery, since ferrous iron can easily be used in the Fenton reaction to produce free radicals such as hydroxyl radicals [28][33]. In this way, the iron can participate directly in the peroxidation of phospholipids (PLOOH) that subsequently will lead to the activation of the whole ferroptosis process. Furthermore, iron is the mineral that catalyzes many important physiological reactions in the cell, such as the formation of most ROS, which can in turn contribute to lipid peroxidation and, thus, to ferroptosis [29][34].
3. Antioxidant Defense
Two years after the first definition of ferroptosis, in 2014, Yang et al. identified GPX4 as a protein playing a crucial role in this mechanism [30][35], although other GPX4-independent pathways were also described. GPX4 is an antioxidant enzyme that can reduce phospholipid hydroperoxide to corresponding alcohols in the cell membrane; therefore, it is considered the key element of ferroptosis inhibition. GPX4 is a selenoprotein that uses reduced glutathione (GSH) as an essential substrate; thus, it is dependent on GSH availability for its proper functioning [31][36] and consists of three amino acids, glutamate, cysteine, and glycine, with a binding that is mediated by glutamate-cysteine ligase and glutathione synthetase. The intracellular level of GSH is strongly regulated by the uptake of cysteine, which is internalized as cystine via the so-called Xc-complex, a glutamate-cystine membrane antiport, consisting of the SLC7A11 (xCT) and SLC3A2 (4F2hc) subunits [32][33][37,38].
The deprivation of cysteine in the cell causes GSH depletion, which can inhibit the GPX4 function, thus leading to the accumulation of lipid hydroperoxides and to ferroptosis due to membrane damage. On the other hand, GPX4-independent pathways were described such as the FSP1 (ferroptosis inhibitor protein 1)-CoQ antioxidant complex that functions only in GPX4-deprived cells. In the membrane, FSP1 reduces ubiquinone to ubiquinol to limit the increase in ROS in the plasma membrane [34][35][39,40].
4. Lipid Metabolism
The lipid metabolism is the third pathway that comes into play to induce ferroptosis through lipid peroxidation. The final products of this process, such as malondialdehyde (MDA), cause membrane instability through damage of the lipid bilayer, which increases the cell permeability and can lead to cell death [32][37].
The major players of this pathway are the Polyunsaturated Fatty Acids (PUFA), because they are more prone to go into peroxidation especially during ferroptosis development.
PUFAs can be synthesized starting from the acyl-CoA synthetase long-chain family member 4 (ACSL4) that binds PUFA to coenzyme A (CoA) to produce acyl-CoA. Afterward, thanks to the action of several acyltransferases, acyl-CoA can be re-esterified in phospholipids [36][37][41,42]. Linoleic acid and arachidonic acid are the most abundant PUFAs in the cell that can be oxidized into hydroperoxides by lipoxygenase (LOX) action. Indeed, LOXs are enzymes that contain the iron element and use it as a cofactor for catalyzing the deoxygenation of both esterified and not esterified PUFAs to produce lipid hydroperoxide in a pro-ferroptosis full picture [6][30][38][11,35,43]. Thus, iron metabolism (i.e., iron levels), antioxidant actions, and lipid metabolism (i.e., phospholipid peroxidation) each have an essential role in triggering critical changes in the cell, leading to cell death through ferroptosis. Regarding the many levels in which ferroptosis can be regulated, the role of non-coding RNAs and especially microRNAs may represent a crucial determinant to finely tune this cell death pathway.