Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Dean Liu and Version 2 by Alina Nishchakova.
Nickel is a well-known catalyst in hydrogenation and dehydrogenation reactions. It is currently used in industrial processes as a homogenous and heterogeneous catalyst. However, to reduce the cost and increase the efficiency of catalytic processes, the development of single-atom catalysts (SACs) seems promising. Some SACs have already shown increased activity and stability as compared to nanoparticle catalysts.
- single-atom catalyst
- nickel
- hydrogenation
- dehydrogenation
1. Introduction
Nickel, the 28th chemical element of Mendeleev’s periodic table of chemical elements, is relatively widespread; its prevalence in the Earth’s crust is 0.009 wt% [1]. The study of the catalytic activity of nickel began in 1879 by the French chemists Paul Sabatier and Jean-Baptiste Senderens, and the reaction that first showed its activity, namely the direct one-stage hydrogenation of ethylene to ethane, is known as the Sabatier-Senderens reaction. Sabatier was the first to widely demonstrate the catalytic activity of nickel, which he described in detail in his famous book “Catalysis in Organic Chemistry” [2]. It seems that many researchers who have studied the catalytic reactions with this particular metal have heard Sabatier’s famous description of a nickel catalyst: “It can be compared to a spirited horse, delicate, difficult to control, and incapable of sustainable work”. However, few of them specify that later in his book, Sabatier determined that the pretreatment of nickel greatly affects its catalytic activity. Sabatier clearly formulated the conditions and features of the catalyst’s preparation, and the reactions of hydrogenation and dehydrogenation of organic compounds served as the starting point for the development of nickel catalysts.
Nowadays, nickel-based catalysts are widely used both at laboratory and industrial scales. Ethylene carbonylation in the presence of nickel carbonyl makes it possible to synthesize propionic acid (Reppe reaction) [3], which, in turn, is widely used in the production of drugs (ibuprofen, etc.), plastics (for example, polyvinyl propionate), surfactants, and also as a preservative E280. The presence of the Ni[(ArO)3P]4 catalyst ensures the hydrocyanation of 1,3–butanediene followed by the isomerization of the reaction products to adiponitrile [4]. The Raney Ni catalyst is used in many hydrogenation reactions [5], for instance, in the conversion of benzene to cyclohexane, in the production of sorbitol from glucose, and in the production of amines from nitro-containing compounds. In the examples given, nickel, with the exception of Raney Ni, acts as a homogeneous catalyst, which is usually inferior to heterogeneous catalysts in industrial processes due to the difficulties associated with separating the catalyst and the reaction mixture. However, homogeneous catalysts have an undeniable advantage since each Ni-containing molecule is a catalytically active center, which allows them to be used in a small concentration, in contrast to heterogeneous catalysts. To offset this shortcoming, the current development of heterogeneous catalysis is focused on single-atom catalysts (SACs).
The development of SACs is a trend of the last decade in modern catalysis, that is just beginning to gain momentum. Many scientists around the world are reporting that the use of catalytically active single atoms improves the efficiency of some reactions by increasing conversion, increasing selectivity to target products, and prolonging the operation of catalysts due to their exceptional stability. Carrying out a catalytic reaction on a specific single atom stabilized by a support makes it possible to reduce the loading of the metal as a whole. Such an approach allows one to reduce overall costs and save non-renewable resources (for example, the content of the most commonly used platinum group metals in the Earth’s crust is only about 10−6–10−8 wt% [1]). The change in catalytic activity is associated with the nature of active sites.
Figure 1 shows how metal-based catalysts can be classified by size, from bulk metal, which does not require the presence of a support, to single atom, which does. In each case presented, the surface atoms can catalyze the reaction; however, the particle surface structure, as well as the total number of metal atoms on the surface, will determine the catalytic activity. SincThe this review research is devoted to nickel, weresearchers should consider its crystal—a face-centered cubic lattice [6]. In this structure, each bulk Ni atom has a coordination number of 12 (saturated coordination), while the surface atoms located in the center of the cube faces are bonded to eight atoms, the atom in the middle of the edge has a coordination number of five, and the nickel atom located at the corner of the lattice is connected to three atoms. Thus, the highest degree of unsaturation is observed for the corner surface atoms, which makes them energetically and often catalytically more attractive. However, this reasoning is applicable only to particles with more than 13 atoms, having at least one internal bond with the maximum saturation, and having the Ni crystal lattice. In the case of amorphous particles or somehow ordered clusters consisting of less than 12 atoms, bond unsaturation for each atom, especially for surface ones, will increase, reaching a maximum for a single atom. Having the highest surface energy, a single Ni atom, like any other metal, needs to be strongly stabilized on the support to prevent its migration for the purpose of binding (for example, agglomeration) in order to reduce the free surface energy. Thus, with the transition from bulk metal to a single atom, there is a loss of stability, often accompanied by an increase in the activity of the catalytic center due to an increase in the unsaturation degree.
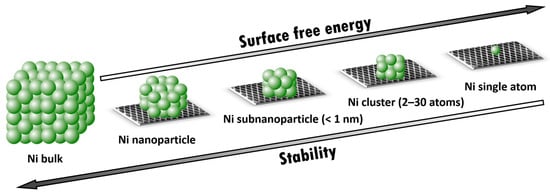
Figure 1. From Ni bulk to supported Ni single atom.
The study of metals in the atomic state is associated with difficulties caused by the resolution of characterization methods. Many reviews are devoted to the characterization of metal in a single-atom state [7,8,9,10,11,12,13,14][7][8][9][10][11][12][13][14]. In the simplest case, indirect confirmation can be used based on a set of methods that confirm the presence of metal in the sample, but its absence in the form of bulk or nanoparticles. However, researchers most often use the three methods depicted: X-ray photoelectron spectroscopy (XPS), electron microscopy, and X-ray absorption fine structure (XAFS) spectroscopy. XPS can be used to determine the electronic state of nickel. Using XAFS spectroscopy, it is possible to determine the nickel oxidation state (X-ray absorption near edge structure, XANES region) and atom environment (Fourier transform of extended X-ray absorption fine structure, EXAFS region). To determine the surrounding atoms, bond lengths, and coordination numbers, it is necessary to carry out additional fitting of the spectra. To directly show the monatomic distribution of the metal, the authors usually use electron microscopy. Thanks to the resolution achieved by modern microscopes, the single metal atoms and the lattice in which they are located can be observed. Thus, by combining the three characterization methods presented, the resulting metal single atoms can be fully investigated, which is necessary to further establish the ‘material-properties-application’ relationship.
Over the past few years, the number of publications devoted to Ni SACs has increased. More than half of them are related to the study of electrocatalytic reactions and corresponding density functional theory (DFT) calculations. This tendency is expected due to humanity’s attempts to avoid the production of energy partially or completely by releasing it from non-renewable, non-ecofriendly oil and gas, while many advanced clean energy technologies require highly active catalysts to lower the energy barrier and increase the reaction rate with an efficient and stable route.
2. Application of Supported Ni Single-Atom Catalysts
In the Introduction section, wresearche rs briefly discussed the historical and modern applications of Ni-based catalysts, which are mainly related to the catalysis of hydrogenation and dehydrogenation processes. When discussing catalysts for such reactions, one cannot fail to mention the competition between palladium and nickel, which was well covered in a recent review by Chernyshov and Ananikov [106][15]. Indeed, being one under the other in the same group of the periodic table, these metals are analogues; however, the difference in the periods causes differences in redox potentials and the availability of different oxidation states, which provides outstanding opportunities for Ni catalysis for complex multistep transformations and the discovery of new catalytic reactions. Thus, wresearchers will consider both the theoretical and practical results of using Ni SACs in various hydrogenation and dehydrogenation reactions. It should be noted that the development of Ni SACs’ application field is not much different from the historical ones for Ni bulk and Ni nanoparticles: this is the breaking of X–H bonds, where X is various atoms, for example, C or O, and H is a hydrogen atom. In this regard, for convenience, weresearchers will further consider separately the activity of heterogeneous catalysts based on Ni single atoms in breaking various X–H bonds, and we researchers will start with the C–H bond.2.1. C–H Activation
Currently, the most common catalysts for the activation of C–H bonds are Ni-based catalysts. The activity of nickel is associated with the presence of an unfilled d orbital, which can accept the σ electron of the C–H bond, thus weakening or breaking it [107][16]. Such an approach to the activation of hydrocarbons can facilitate cross-coupling reactions, leveling the disadvantages known for this reaction: the use of an expensive catalyst (Pd-based), the presence of several stages of pre-functionalization, which entails a multi-stage reaction, and the presence of by-products. However, the activation of the C–H bond also has its drawbacks, in particular, high reaction temperature and high metal loading are needed. The best-known nickel-catalyzed reaction involving C–H activation is the dry reforming of methane (DRM).Dry Reforming of Methane
DRM is a process for the simultaneous synthesis of H2 and CO (syngas) via the conversion of carbon dioxide and methane. Even though this reaction may not actually involve hydrogenation and dehydrogenation, its mechanism includes the C–H bond cleavage as an important step of the reaction. The general reaction (1) describing this process is endothermic with a standard enthalpy of 247 kJ mol–1 and, therefore, requires high temperatures (>500 °C) [59,60,108][17][18][19].CH4+CO2→2CO+2H2
CH4(g)+cat→CH4*
CH4*→CH3*+H*
2CO→CO2+C
CH4→C+2H2
2.2. H–H Activation
Hydrogen is the simplest molecule, and its properties are the most studied. Since this pure resource is available in abundance at a very low cost, catalytic hydrogenation is a mainstream technology in both research and industry. Molecular hydrogen is not very active under ambient conditions, but many positively charged transition metal atoms are capable of bonding and activating H2. As is well known, bulk Ni and Pd have a very strong hydrogen adsorption affinity, can easily form metal hydrides, and are widely considered good catalysts due to their excellent hydrogen solubility, corrosion resistance, and diffusivity. As indicated in the literature, the adsorption energy of the hydrogen molecule on the Ni single atom [128,129][39][40] is higher than that on bulk Ni [130][41]. The initiation of the hydrogenation reaction most often occurs with the dissociation of the hydrogen molecule. Homolytic decomposition yields adsorbed H* atoms, while heterolytic decomposition produces partially charged Hδ+ and Hδ– species. The type of decomposition, the energy barrier, and the overall energy profile of the reaction strongly depend on the structure of the catalytically active Ni site. For example, according to DFT calculations of a catalytically active Ni atom coordinated to three and four carbon atoms in single and double graphene vacancies, H2 dissociation is an endothermic process with energy barriers of 0.69 and 0.33 eV, respectively [131][42]. As can be seen, the dissociation of the adsorbed H2 is an energy-consuming process [128,131,132][39][42][43]. However, with an increase in the number of Ni atoms to three in a catalytically active site, the dissociation is almost barrier-free [130][41]. Interestingly, a similar effect was observed when considering the reverse reaction, where the recombination of hydrogen atoms into a molecule took place [133][44]. After decomposition, hydrogen atoms can spontaneously migrate to support atoms. This effect is called the hydrogen ‘spillover’ process, and it may take place in the metal-carbon support system [134][45]. Such behavior, in particular, was observed for some nitrogen-containing systems [128][39] and also for MoS2 [132][43]. For instance, the addition of Ni single-atoms on MoS2 support causes an excess of electron density on the nearest sulfur atoms, thus enhancing their activity towards hydrogen adsorption [32,49][46][47]. Although the activation of a hydrogen molecule can also occur due to an increase in the H–H bond length after adsorption onto the Ni atom. Furthermore, during the co-adsorption of the reagents on the metal atom, the breaking of this bond for the further course of the reaction will be easier.2.3. O–H Activation2.3. O–H Activation
The use of hydrogen as a hydrogenating agent has some disadvantages associated with its gaseous state under ambient conditions. In the reactions discussed above, the typical hydrogen pressure was in the range of 1–3 MPa, which introduces difficulties in carrying out reactions both on laboratory and industrial scales. To overcome this, other molecules are used from which the hydrogen atom can be obtained, such as alcohols. Wang et al. studied the effect of the composition and structure of the Ni–NxCy site in graphene on the adsorption of isopropyl alcohol and the subsequent detachment of hydrogen from it [149][48]. The Ni–N4 and Ni–N3 sites turned out to be the most energetically favorable. It was shown that, despite the energetic preference for its formation, the flat structure of the Ni–N4 site sterically prevents the dissociation of the O–H bond and makes the process highly endothermic. On the contrary, the Ni–N3 site, in which the Ni atom protrudes above the graphene surface, not only easily adsorbs the isopropyl alcohol molecule due to the overlapping of the p-orbitals of the O atom and the d-orbitals of the Ni atom but also easily breaks the O–H bond.2.4. N–H Activation
In addition to the reactions of the formation of hydrogen atoms or molecules by activation of C–H, H–H, and O–H bonds, which are widely covered in the literature, some authors considered the possibility of using single-atom Ni sites for activation of other bonds, for example, N–H. Feng et al. studied the mechanism of reduction of Br–C6H5–NO2 to Br–C6H5–NH2 at Ni–N4 and Ni–N3 sites using N2H4·H2O as a hydrogen donor [155][49]. Since the former catalytically active site exhibited significantly higher activity than the second one, the energy values given below correspond to the Ni–N3 site. Hydrazine was adsorbed with the formation of the Ni–N bond, and the N–H bond breaking energy was equal to 0.93 eV, with both N2H3* and H* species remaining coordinated to the nickel atom. The subsequent adsorption of the R–NO2 molecule was exothermic. The limiting stage of the reaction was the chemo-selective reduction process of the −NOH group through the recombination of the adsorbed hydrogen atom and the hydrogen atom of the −OH group—with an activation energy of 0.96 eV. The presence of the Br substituent did not adversely affect the reduction of the nitro group; moreover, the authors calculated that 1.18 eV would be required to break the C–Br bond, which makes this process kinetically unfavorable. In practice, the catalyst achieved 100% conversion and selectivity at 60 °C within 3 h of operation, and the calculated TOF was higher than that for the catalyst containing Ni nanoparticles. It is interesting that theoretical calculations of the decomposition of N2H4 at the Ni–C3 site did not show such good results as in the work [156][50]. The adsorption of the molecule on the catalytically active Ni atom still occurred due to the overlapping of the 3d and 2p orbitals of Ni and N, respectively. However, the authors calculated not only the dissociation of the N–H bond but also the dissociation of the N–N bond and showed that the second way is energetically preferable (0.86 and 0.51 eV, respectively). Moreover, the final state for the system after the N–H bond cleavage was endothermic, while after the N–N bond cleavage, it was exothermic, which indicates the potential unsuitability of the Ni–C3 site as catalytically active in the production of H* from hydrazine. Although this site still showed potential activity and high selectivity in the formation of ammonia.References
- Abundance in Earth’s Crust for All the Elements in the Periodic Table. Available online: https://periodictable.com/Properties/A/CrustAbundance.html (accessed on 2 May 2023).
- Sabatier, P. Catalysis in Organic Chemistry; D. Van Nostrand Company: New York, NY, USA, 1922.
- Keim, W. Nickel: An Element with Wide Application in Industrial Homogeneous Catalysis. Angew. Chem. Int. Ed. Engl. 1990, 29, 235–244.
- Schneider, C.; Leischner, T.; Ryabchuk, P.; Jackstell, R.; Junge, K.; Beller, M. Development of Bulk Organic Chemical Processes—History, Status, and Opportunities for Academic Research. CCS Chem. 2021, 3, 512–530.
- Sun, Z.; Zhang, Z.-H.; Yuan, T.-Q.; Ren, X.; Rong, Z. Raney Ni as a Versatile Catalyst for Biomass Conversion. ACS Catal. 2021, 11, 10508–10536.
- Jette, E.R.; Foote, F. Precision Determination of Lattice Constants. J. Chem. Phys. 1935, 3, 605–616.
- Kaiser, S.K.; Chen, Z.; Faust Akl, D.; Mitchell, S.; Pérez-Ramírez, J. Single-Atom Catalysts across the Periodic Table. Chem. Rev. 2020, 120, 11703–11809.
- Shang, Y.; Duan, X.; Wang, S.; Yue, Q.; Gao, B.; Xu, X. Carbon-Based Single Atom Catalyst: Synthesis, Characterization, DFT Calculations. Chin. Chem. Lett. 2022, 33, 663–673.
- Qi, P.; Wang, J.; Djitcheu, X.; He, D.; Liu, H.; Zhang, Q. Techniques for the Characterization of Single Atom Catalysts. RSC Adv. 2022, 12, 1216–1227.
- Wu, J.; Xiong, L.; Zhao, B.; Liu, M.; Huang, L. Densely Populated Single Atom Catalysts. Small Methods 2020, 4, 1900540.
- Yang, J.; Li, W.; Wang, D.; Li, Y. Electronic Metal–Support Interaction of Single-Atom Catalysts and Applications in Electrocatalysis. Adv. Mater. 2020, 32, 2003300.
- Qi, K.; Chhowalla, M.; Voiry, D. Single Atom Is Not Alone: Metal–Support Interactions in Single-Atom Catalysis. Mater. Today 2020, 40, 173–192.
- Kottwitz, M.; Li, Y.; Wang, H.; Frenkel, A.I.; Nuzzo, R.G. Single Atom Catalysts: A Review of Characterization Methods. Chemistry–Methods 2021, 1, 278–294.
- Liu, J.; Bunes, B.R.; Zang, L.; Wang, C. Supported Single-Atom Catalysts: Synthesis, Characterization, Properties, and Applications. Environ. Chem. Lett. 2018, 16, 477–505.
- Chernyshev, V.M.; Ananikov, V.P. Nickel and Palladium Catalysis: Stronger Demand than Ever. ACS Catal. 2022, 12, 1180–1200.
- Yang, Y.; Li, W.; Xu, H. A New Explanation for the Carbon Deposition and Elimination over Supported Ni, Ni-Ce and Ni-Co Catalysts for CO2-Reforming of Methane. React. Kinet. Catal. Lett. 2002, 77, 155–162.
- Tang, Y.; Wei, Y.; Wang, Z.; Zhang, S.; Li, Y.; Nguyen, L.; Li, Y.; Zhou, Y.; Shen, W.; Tao, F.F.; et al. Synergy of Single-Atom Ni1 and Ru1 Sites on CeO2 for Dry Reforming of CH4. J. Am. Chem. Soc. 2019, 141, 7283–7293.
- Akri, M.; Zhao, S.; Li, X.; Zang, K.; Lee, A.F.; Isaacs, M.A.; Xi, W.; Gangarajula, Y.; Luo, J.; Ren, Y.; et al. Atomically Dispersed Nickel as Coke-Resistant Active Sites for Methane Dry Reforming. Nat. Commun. 2019, 10, 5181.
- Vogt, C.; Kranenborg, J.; Monai, M.; Weckhuysen, B.M. Structure Sensitivity in Steam and Dry Methane Reforming over Nickel: Activity and Carbon Formation. ACS Catal. 2020, 10, 1428–1438.
- Khairudin, N.F.; Sukri, M.F.F.; Khavarian, M.; Mohamed, A.R. Understanding the Performance and Mechanism of Mg-Containing Oxides as Support Catalysts in the Thermal Dry Reforming of Methane. Beilstein J. Nanotechnol. 2018, 9, 1162–1183.
- Baharudin, L.; Rahmat, N.; Othman, N.H.; Shah, N.; Syed-Hassan, S.S.A. Formation, Control, and Elimination of Carbon on Ni-Based Catalyst during CO2 and CH4 Conversion via Dry Reforming Process: A Review. J. CO2 Util. 2022, 61, 102050.
- Wei, J.; Iglesia, E. Isotopic and Kinetic Assessment of the Mechanism of Reactions of CH4 with CO2 or H2O to Form Synthesis Gas and Carbon on Nickel Catalysts. J. Catal. 2004, 224, 370–383.
- Lustemberg, P.G.; Ramírez, P.J.; Liu, Z.; Gutiérrez, R.A.; Grinter, D.G.; Carrasco, J.; Senanayake, S.D.; Rodriguez, J.A.; Ganduglia-Pirovano, M.V. Room-Temperature Activation of Methane and Dry Re-Forming with CO2 on Ni-CeO2 (111) Surfaces: Effect of Ce3+ Sites and Metal–Support Interactions on C–H Bond Cleavage. ACS Catal. 2016, 6, 8184–8191.
- Pantha, N.; Ulman, K.; Narasimhan, S. Adsorption of Methane on Single Metal Atoms Supported on Graphene: Role of Electron Back-Donation in Binding and Activation. J. Chem. Phys. 2020, 153, 244701.
- Wu, J.; Gao, J.; Lian, S.; Li, J.; Sun, K.; Zhao, S.; Kim, Y.D.; Ren, Y.; Zhang, M.; Liu, Q.; et al. Engineering the Oxygen Vacancies Enables Ni Single-Atom Catalyst for Stable and Efficient C-H Activation. Appl. Catal. B Environ. 2022, 314, 121516.
- Akri, M.; El Kasmi, A.; Batiot-Dupeyrat, C.; Qiao, B. Highly Active and Carbon-Resistant Nickel Single-Atom Catalysts for Methane Dry Reforming. Catalysts 2020, 10, 630.
- Bellido, J.D.A.; Assaf, E.M. Effect of the Y2O3–ZrO2 Support Composition on Nickel Catalyst Evaluated in Dry Reforming of Methane. Appl. Catal. Gen. 2009, 352, 179–187.
- Paladino Lino, A.V.; Rodella, C.B.; Assaf, E.M.; Assaf, J.M. Methane Tri-Reforming for Synthesis Gas Production Using Ni/CeZrO2/MgAl2O4 Catalysts: Effect of Zr/Ce Molar Ratio. Int. J. Hydrog. Energy 2020, 45, 8418–8432.
- Wang, N.; Yu, X.; Shen, K.; Chu, W.; Qian, W. Synthesis, Characterization and Catalytic Performance of MgO-Coated Ni/SBA-15 Catalysts for Methane Dry Reforming to Syngas and Hydrogen. Int. J. Hydrog. Energy 2013, 38, 9718–9731.
- Zuo, Z.; Liu, S.; Wang, Z.; Liu, C.; Huang, W.; Huang, J.; Liu, P. Dry Reforming of Methane on Single-Site Ni/MgO Catalysts: Importance of Site Confinement. ACS Catal. 2018, 8, 9821–9835.
- Juan-Juan, J.; Román-Martínez, M.C.; Illán-Gómez, M.J. Effect of Potassium Content in the Activity of K-Promoted Ni/Al2O3 Catalysts for the Dry Reforming of Methane. Appl. Catal. Gen. 2006, 301, 9–15.
- Zapata, B.; Valenzuela, M.A.; Palacios, J.; Torres-Garcia, E. Effect of Ca, Ce or K Oxide Addition on the Activity of Ni/SiO2 Catalysts for the Methane Decomposition Reaction. Int. J. Hydrog. Energy 2010, 35, 12091–12097.
- Kim, S.M.; Abdala, P.M.; Margossian, T.; Hosseini, D.; Foppa, L.; Armutlulu, A.; van Beek, W.; Comas-Vives, A.; Copéret, C.; Müller, C. Cooperativity and Dynamics Increase the Performance of NiFe Dry Reforming Catalysts. J. Am. Chem. Soc. 2017, 139, 1937–1949.
- Al-Fatesh, A. Suppression of Carbon Formation in CH4–CO2 Reforming by Addition of Sr into Bimetallic Ni–Co/γ-Al2O3 Catalyst. J. King Saud Univ. Eng. Sci. 2015, 27, 101–107.
- Alipour, Z.; Rezaei, M.; Meshkani, F. Effect of Alkaline Earth Promoters (MgO, CaO, and BaO) on the Activity and Coke Formation of Ni Catalysts Supported on Nanocrystalline Al2O3 in Dry Reforming of Methane. J. Ind. Eng. Chem. 2014, 20, 2858–2863.
- Roh, H.-S.; Jun, K.-W. Carbon Dioxide Reforming of Methane over Ni Catalysts Supported on Al2O3 Modified with La2O3, MgO, and CaO. Catal. Surv. Asia 2008, 12, 239–252.
- Ma, Q.; Sun, J.; Gao, X.; Zhang, J.; Zhao, T.; Yoneyama, Y.; Tsubaki, N. Ordered Mesoporous Alumina-Supported Bimetallic Pd–Ni Catalysts for Methane Dry Reforming Reaction. Catal. Sci. Technol. 2016, 6, 6542–6550.
- Huang, S.; Xu, H.; Li, H.; Guo, Y.; Sun, Z.; Du, Y.; Li, H.; Zhang, J.; Pang, R.; Dong, Q.; et al. Preparation and Characterization of Char Supported NiCu Nanoalloy Catalyst for Biomass Tar Cracking Together with Syngas-Rich Gas Production. Fuel Process. Technol. 2021, 218, 106858.
- Wang, Y.; Kang, L. Selective Hydrogenation of Acetylene Catalyzed by Nickel and Nitrogen-Doped C34: A Density Functional Theory Study. Chem. Phys. Lett. 2020, 757, 137871.
- Poldorn, P.; Wongnongwa, Y.; Mudchimo, T.; Jungsuttiwong, S. Theoretical Insights into Catalytic CO2 Hydrogenation over Single-Atom (Fe or Ni) Incorporated Nitrogen-Doped Graphene. J. CO2 Util. 2021, 48, 101532.
- Xue, M.; Jia, J.; Wu, H. A Density Functional Theory Study on the Catalytic Performance of Metal (Ni, Pd) Single Atom, Dimer and Trimer for H2 Dissociation. Chem. Phys. 2022, 552, 111336.
- Zhuo, H.-Y.; Yu, X.; Yu, Q.; Xiao, H.; Zhang, X.; Li, J. Selective Hydrogenation of Acetylene on Graphene-Supported Non-Noble Metal Single-Atom Catalysts. Sci. China Mater. 2020, 63, 1741–1749.
- Sun, M.; Nelson, A.; Adjaye, J. Ab Initio DFT Study of Hydrogen Dissociation on MoS2, NiMoS, and CoMoS: Mechanism, Kinetics, and Vibrational Frequencies. J. Catal. 2005, 233, 411–421.
- Bing, Q.; Liu, W.; Yi, W.; Liu, J. Ni Anchored C2N Monolayers as Low-Cost and Efficient Catalysts for Hydrogen Production from Formic Acid. J. Power Sources 2019, 413, 399–407.
- Prins, R. Hydrogen Spillover. Facts and Fiction. Chem. Rev. 2012, 112, 2714–2738.
- Wang, Q.; Zhao, Z.L.; Dong, S.; He, D.; Lawrence, M.J.; Han, S.; Cai, C.; Xiang, S.; Rodriguez, P.; Xiang, B.; et al. Design of Active Nickel Single-Atom Decorated MoS2 as a PH-Universal Catalyst for Hydrogen Evolution Reaction. Nano Energy 2018, 53, 458–467.
- Zhang, H.; Yu, L.; Chen, T.; Zhou, W.; Lou, X.W.D. Surface Modulation of Hierarchical MoS2 Nanosheets by Ni Single Atoms for Enhanced Electrocatalytic Hydrogen Evolution. Adv. Funct. Mater. 2018, 28, 1807086.
- Wang, F.-F.; Guo, R.; Jian, C.; Zhang, W.; Xue, R.; Chen, D.-L.; Zhang, F.; Zhu, W. Mechanism of Catalytic Transfer Hydrogenation for Furfural Using Single Ni Atom Catalysts Anchored to Nitrogen-Doped Graphene Sheets. Inorg. Chem. 2022, 61, 9138–9146.
- Feng, B.; Guo, R.; Cai, Q.; Song, Y.; Li, N.; Fu, Y.; Chen, D.-L.; Zhang, J.; Zhu, W.; Zhang, F. Construction of Isolated Ni Sites on Nitrogen-Doped Hollow Carbon Spheres with Ni–N3 Configuration for Enhanced Reduction of Nitroarenes. Nano Res. 2022, 15, 6001–6009.
- Genç, A.E.; Küçük, H.; Alp, I.O.; Akça, A. Hydrazine Decomposition on Nickel-Embedded Graphene. Int. J. Hydrog. Energy 2020, 45, 33407–33418.
More