Cancer Stem Cells (CSCs) is a subset of cancer cells with the ability to self-renew and to differentiate into non-CSC cancer cells within the tumor mass. The CSC field was shaped by great research done on hematopoietic stem cells (HSCs). HSCs are hierarchically arranged with HSCs being the founder cells that undergo asymmetric cell division giving rise to differentiated daughter cells and one quiescent stem cell with self-renewal abilities. CSCs are a subpopulation of cancer cells known to be resistant to therapy and cause metastasis. CSCs have been characterized in many cancers with data illustrating that CSCs display great abilities to self-renew, resist therapies due to enhanced epithelial to mesenchymal (EMT) properties, enhanced expression of ATP-binding cassette (ABC) membrane drug transporters, activation of several survival signaling pathways and increased immune evasion as well as DNA repair mechanisms. CSCs also display great heterogeneity with the consequential lack of specific CSC markers presenting a great challenge to their targeting.
- Cancer stem cells
- Drug response
- Chemotherapy
- Radiotherapy
- Drug efflux pumps
- Tumor Microenvironment
- CSC Niche
- EMT
- Metabolism
- Immunotherapy
1. Introduction
The dividing daughter cells will over time become restricted in terms of lineages it can form. The studies on HSCs ignited research on mammalian tissue and cell renewal as well as in cancer. In addition, cancer patients with chronic myeloid leukemia (CML) were shown to have rare quiescent cells also referred to as Philadelphia chromosome-positive and BCR-ABL-positive cells and these cells were able to resist drug treatment [1][2]. The above-mentioned studies and revelations allowed further research on self-renewal and eventually gave birth to the CSC field as it is today. CSCs are able to reproduce primary tumor heterogeneity as well as metastases in distant tissues and organs [3]. As postulated by Paget, cancer cells can escape the primary tumor site and spread to other tissues and organs where they can proliferate and therefore act as “seeds” for the growth of secondary tumors [3]. It is possible that cancer cells can detach from the primary tumor and enter circulation, however, they are likely not to survive the arduous journey to other organs and cannot “seed” metastases at secondary sites. With their demonstrable survival abilities, enhanced expression of transmembrane transporters and tumorigenic abilities, CSCs on the other hand are likely to survive in circulation and be able to “seed” new tumors at secondary sites [4][5]. CSCs are also responsible for the development of therapy resistance, with many studies demonstrating that CSCs are able to withstand conventional therapies such as chemotherapy and radiotherapy [6]. The ability to resist conventional therapies has been attributed to many properties including increased expression of drug transporters, maintenance of a slow dividing state (quiescence) as well as efficient DNA repair mechanisms [7][8][9]. To overcome CSC resistance, new therapies are under development including epigenetic therapies, immunotherapy as well as drugs targeting angiogenesis [10].
From the early days of their discovery, many studies have shown that CSCs are undifferentiated tumor cells able to generate tumors [11][12][13]. To date, several studies have been able to prove the existence of CSCs in cancers such as CML, ovarian, lung and breast cancer [14][15]. Methods used to identify CSCs range from antibody-based isolation, enzyme activity of ALDH, tumorsphere formation, use of dyes such as PKH26 and side population sorting [16][17]. Side population cells display enhanced abilities to efflux dyes and drugs at a higher rate than the main cell population due to increased expression of ATP-binding cassette (ABC) transporter proteins. These methods are all not specific, and in most cases, scientists combine these methods to get a cell population with high numbers of CSCs. The gold standard method to study whether cancer cells have tumor-initiating capabilities is the use of limiting dilution in xenograft animals. A detailed review of CSCs definition and terminology is provided by Valent and colleagues [6]. Recently introduced “humanized” animal models are better models than traditional animal models as they can recapitulate some human cancers better [18][19].
Due to their ability to resist therapy, CSCs can travel to distant sites and form new tumors. Whilst the process of metastasis appears disorganized, metastatic lesions are the main cause of cancer deaths and therapy resistance [20]. Signaling pathways upregulated and dysregulated in CSCs and CSC-cell interactions are therefore some of the targets of new drugs under development. Conventional cancer treatment strategies mainly target rapidly proliferating cancer cells and can reduce tumor mass, tumor relapse can result from a few remaining cancer cells including CSCs (Figure 1) [21]. Researchers' ability to target CSCs largely depends on new evidence and in-depth characterization of these cells. It is plausible to postulate that long-lasting cancer treatment efficacy can only come from both shrinkage of the primary tumor as well as the prevention of cells such as CSCs from metastasizing to new sites throughout the body.
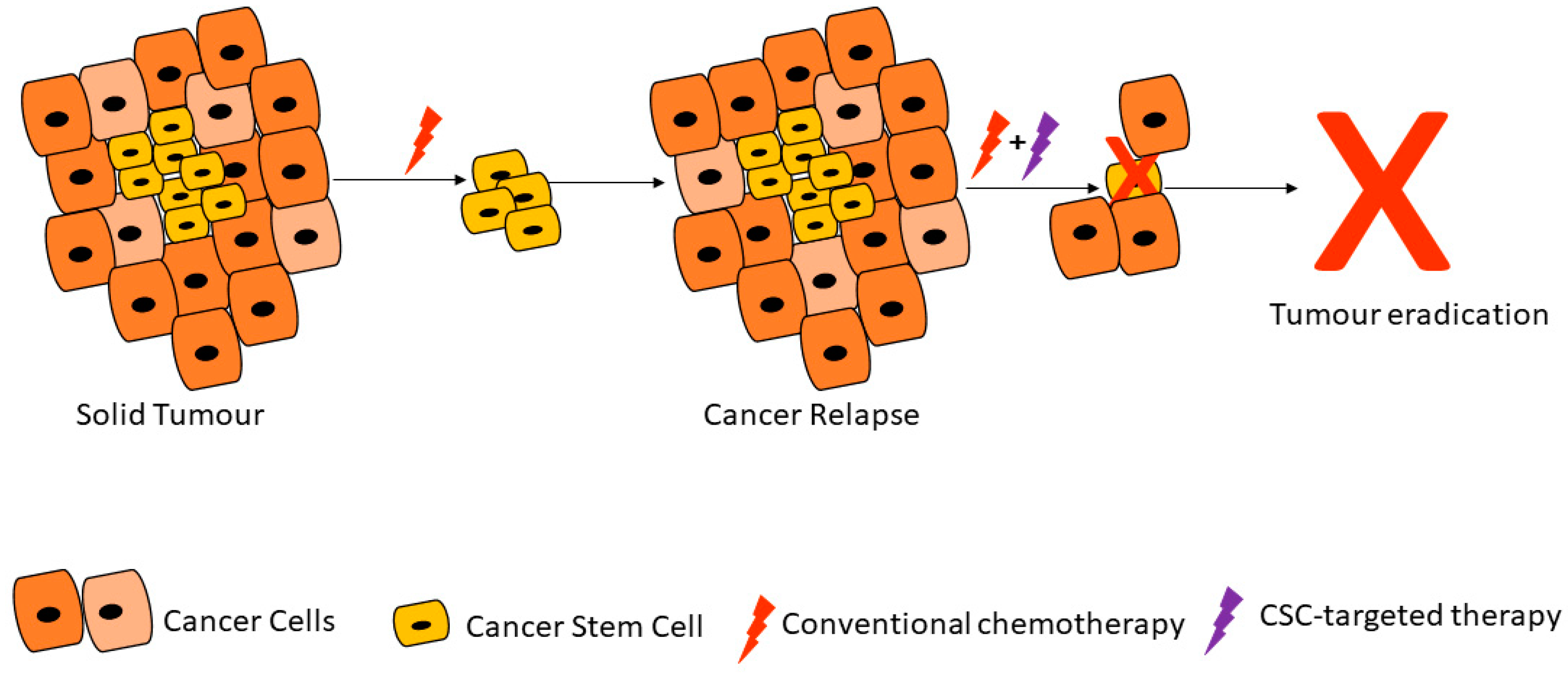
Figure 1. Cancer stem cells are able to resist conventional therapies and form new tumors, unless targeted by cancer stem cell (CSC)-specific therapy. Adapted from Dzobo et al. [21].
2. Properties of Cancer Stem Cells
2.1. Cancer Stem Cell Markers and Therapy Resistance
Current therapies are unable to eliminate cancer partly due to CSCs’ enhanced ability to withstand treatment regimens [6][21]. CSCs are thought to account for a small percentage of the total number of cancer cells within a tumor but have self-renewal and differentiation capabilities [12]. A major hurdle faced by scientists working with CSCs has been the isolation and characterization of these cells. Antibodies against several CSC markers have been used to isolate CSCs from solid tumors [17]. Commonly used CSC markers and methods for isolation and characterization include CD24, CD44, CD133 and ALDH enzymatic assay (Figure 2; Table 1Table 2) [22]. These CSC markers are either used alone or in different combinations in different cancers. For example, gastric CSCs display high CD44, CD133 as well as Lgr5 [23]. Lung CSCs express several markers including CD133+, ALDH1+ and CD44+ [24]. Whilst the same CSC markers can be found in different cancers, some cancers have distinct markers for example melanoma CSCs are ABCB5+ whilst medulloblastoma CSCs are CD15+ (Table 1).
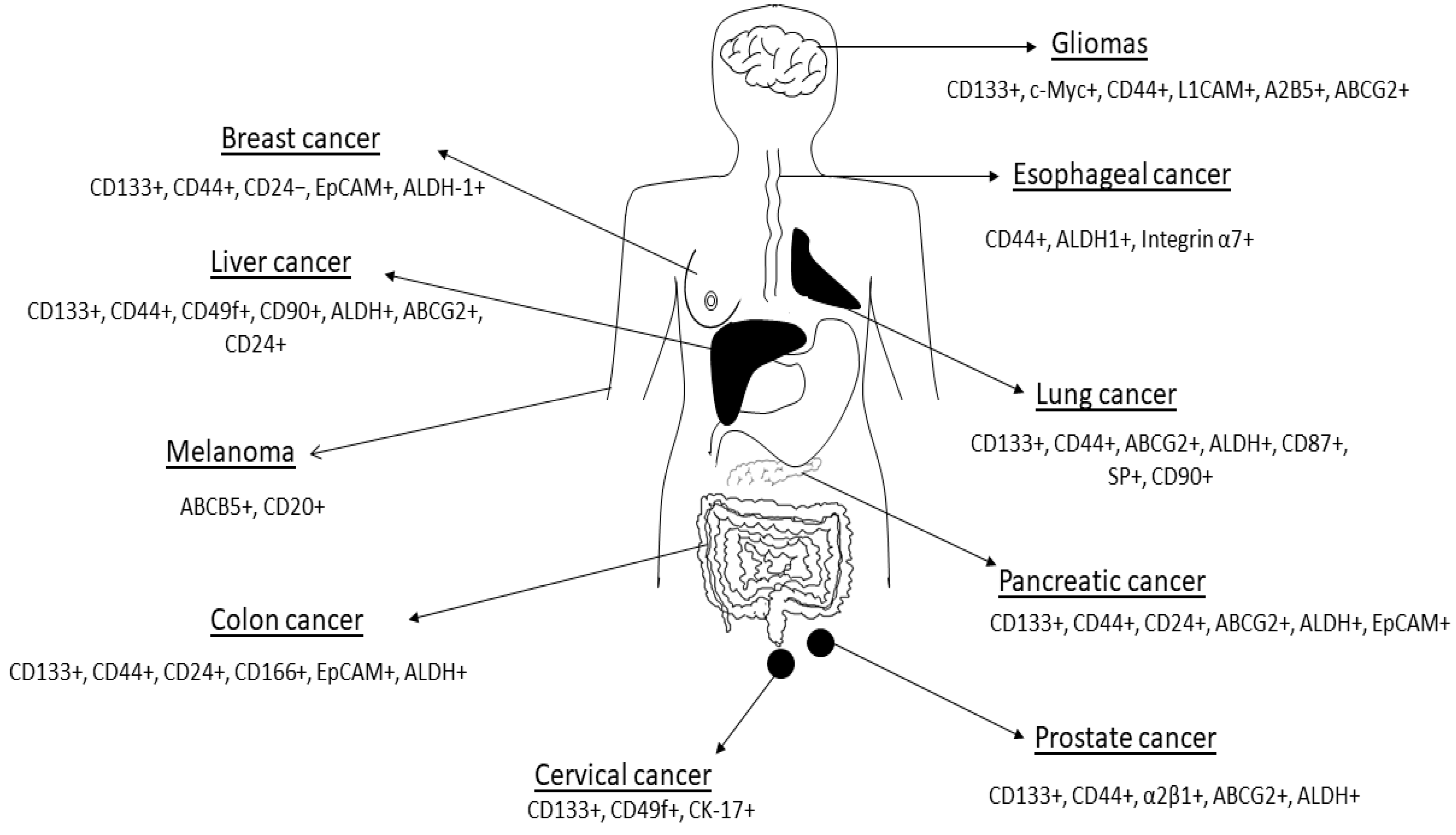
Figure 2. Cancer stem cell markers expressed in some human cancers are shown in the figure. Figure adapted from Dzobo et al. [25]. See Table 1 Table 1 for references. The list of CSC markers is not exhaustive. The CSC markers continue to be refined based on new data.
Table 1. CSC markers expressed in different human cancers *.
| Cancer | CSC Markers | References |
|---|---|---|
| Cervical | CD133+, CD49f+, CK-17+ | [26][27][28] |
| Esophageal | CD44+, ALDH1+, Integrin α7+ | [29][30] |
| Kidney | CD24-, CD44+, CD105+, CD133+ | [31][32][33] |
| Lung cancer | CD44+, CD90+, CD133+, ABCG2+, ALDH+ | [15][24] |
| Colon cancer | CD24+, CD44+, CD133+, EpCAM+, ALDH+ | [34][35][36][37] |
| Liver cancer | CD24+, CD44+, CD90+, CD133+, ALDH+, ABCG2+ | [38][39] |
| Breast cancer | CD24-, CD44+, CD133+, ALDH-1+ | [40][41][42] |
| Gastric | CD44+, CD133+ | [43][44][45] |
| Glioma | CD44+, CD133+, A2B5+, BCRP1+, SSEA-1+ | [46][47] |
| Leukemia (AML) | CD34+, CD38−, CD123+ | [48][49][50] |
| Leukemia (CML) | CD25+, CD26+, CD44+, CD93+, IL1RAP+ | [51][52] |
| Ovarian | CD44+, CD117+, CD133+, ALDH1+ | [53][54] |
| Prostate cancer | CD44+, CD133+, α2β1+, ALDH+ | [55][56][57] |
| Pancreatic cancer | CD44+, CD133+, ABCG2+, ALDH+, EpCAM+ | [58][59][60] |
| Melanoma | ABCB5+, CD20+ | [61][62] |
| Head and neck cancer | CD44+, CD133+ | [63][64] |
| Sarcoma | CD29+, CD117+, CD133+, Nestin+, Stro-1+ | [65][66] |
* The list of CSC markers is not exhaustive. The CSC markers continue to be refined based on new data.
Several studies demonstrated an increase in CSCs in tumors after cancer treatment, clearly illustrating their persistence during treatment [67][68][69]. CSCs are able to resist therapeutic interventions due to several reasons including their cellular plasticity, enhanced expression of ABC drug transporters, ability to detoxify of drugs and compounds, increased adaptation to stressful conditions such as hypoxia, attaining quiescence and activation of survival pathways [69][70][71].
CSCs ability to resist therapy is widespread and referred to as multidrug resistance. This capability stems from the ability of CSCs to express increased detoxifying enzymes, increased activation of survival signaling pathways, DNA repair mechanisms as well as drug efflux pumps [21][70]. In addition, CSCs have been noted for their immune evasion capabilities, their ability to undergo epithelial to EMT as well as to adapt their metabolism to survive low nutrient conditions [69][70]. Thus, the hallmarks of CSCs include quiescence, increased expression of drug metabolizing and detoxifying enzymes, enhanced DNA reparability, the ability to undergo EMT and overexpression of ABC membrane transporters. Lately, CSCs have also been shown to undergo epigenetic reprogramming, making them very difficult to eradicate in cancers [72].
The ALDH superfamily is a large family of proteins and several members including aldehyde dehydrogenase 1 (ALDH1) have been implicated in drug detoxifying activities [73][74]. In its entirety, the ALDH superfamily is composed of 19 enzymes with ALDH1 being the main isoform [73][74][75]. This family of detoxifying enzymes is involved in the oxidation of aldehydes to carboxylic acids as well as retinol to retinoic acid [76][77]. Besides being expressed by normal cells, ALDH1 is expressed highly in CSCs [78][79]. As a result, ALDH1 expression and activity can be used reliably to identify CSCs in some cancers. Vogler and colleagues demonstrated that ALDH1 expression can be used as an independent prognostic marker for low survival in colorectal patients [80]. In addition, van den Hoogen and coworkers also showed that enhanced ALDH1 activity can be used to identify tumor-forming cells as well as cells with the propensity to form prostate cancer metastases [81]. Ueda and colleagues also showed that ALDH1 activity can be used to identify cancer cells with CSC-like properties in human renal cell carcinoma cell line [82]. Ginestier and colleagues demonstrated that ALDH1 is highly expressed in breast CSCs and is a predictor of poor clinical outcome [83]. In addition, ALDH1-expressing cells were able to form xenograft tumors easily [83]. Several other studies demonstrated the successful transplantation of ALDH1-expressing cells into mice [84][85]. The expression of ALDH1 by normal stem cells may explain the aberrant expression of this enzyme in CSCs as normal stem cells are a potential source of CSCs, among other cells [86]. Furthermore, ALDH1 expression has been shown to allow CSCs to resist conventional therapy including commonly used drugs such as paclitaxel, gemcitabine and cisplatin [87][88]. In agreement with the above, several studies demonstrated that inhibition of ALDH1 activity in CSCs sensitizes these cells to several drugs, linking ALDH1 with therapy resistance [89][90].
In addition, CSCs demonstrate increased expression of drug effluxing proteins such as the ABC transporters (Figure 3) [91][92][93]. The ABC family of transporters consists of 49 molecules using ATP as an energy source during the trafficking of proteins across the cell membrane. Many studies have been performed on the characterization of members of this family including ABCB1 (multidrug resistance 1 (MDR1)), ABCG2, ABCC1 and ABCB5 [94][95]. Through elaborate experiments, several research groups demonstrated that CSCs aberrantly express ABC transporters and are able to withstand toxic levels of drugs and other toxins [96][97]. In elaborate experiments performed by Wright and colleagues, the researchers demonstrated that ABCB1 was aberrantly overexpressed in breast CSCs causing resistance to conventional chemotherapy such as paclitaxel and doxorubicin [98]. Frank and coworkers demonstrated that ABCB5 was overexpressed and caused resistance to doxorubicin in CD133+ circulating melanoma cells [99]. Through the use of a monoclonal antibody against ABCB5, the authors were able to induce cancer cell sensitivity to drugs such as doxorubicin [99]. Shi and colleagues demonstrated that ABCG2-expressing CSCs isolated from hepatocellular carcinoma cell lines via the side population technique are able to resist cisplatin and 5-fluorouracil [100]. The above studies and others demonstrated that inhibition of ABC transporters is a potential mechanism of overcoming CSC chemoresistance [101][102]. Several studies have been performed on the inhibition of ABC transporters and have shown remarkable success in sensitizing both cancer cells and CSCs to several drugs [103][104]. For example, Marcelletti and colleagues utilized zosuquidar, an inhibitor of P-gp (ABCB1) to sensitize cancer cells in acute myeloid leukemia [103][104].
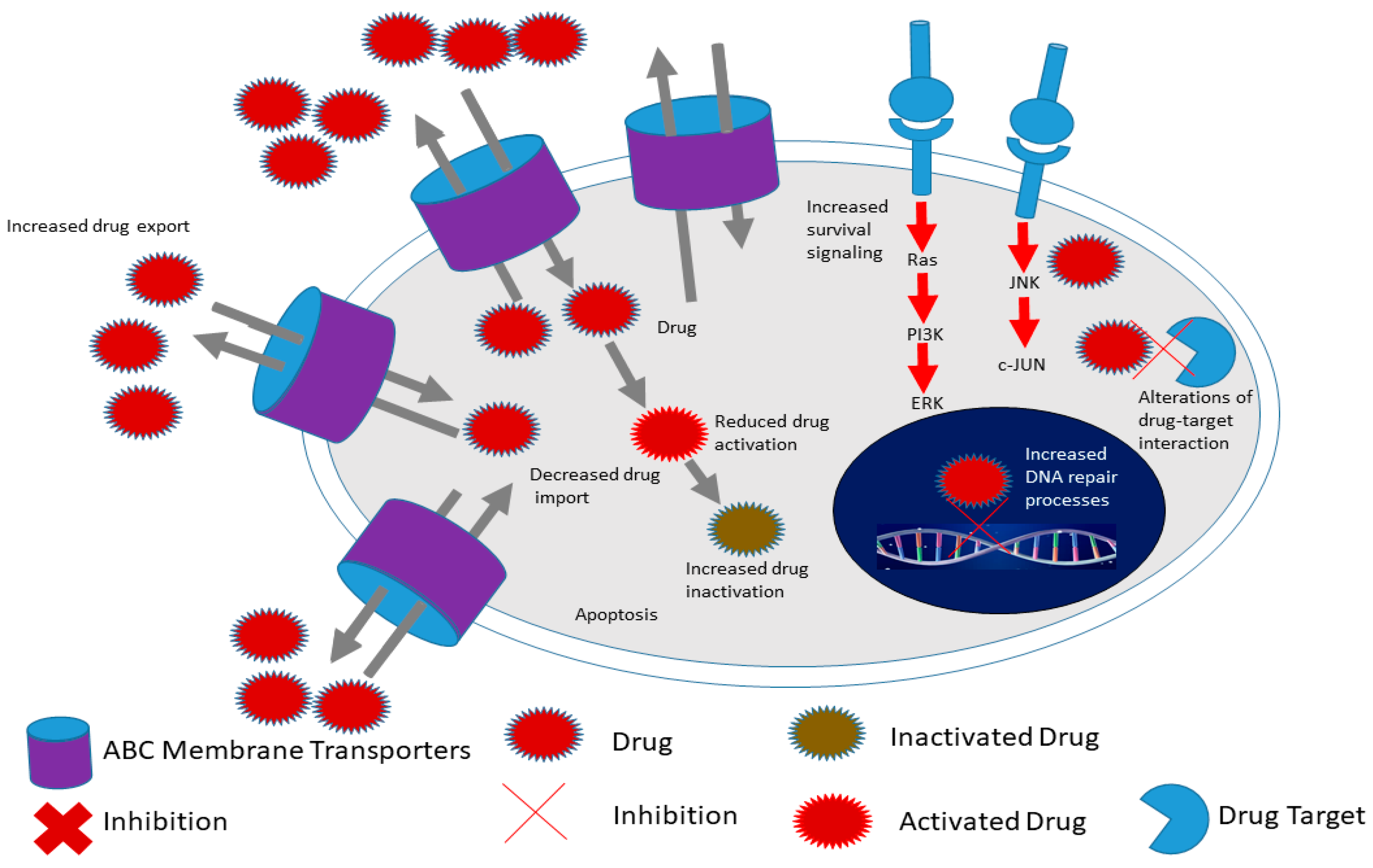
Figure 3. Hallmarks of cancer stem cells include increased expression of ATP-binding cassette (ABC) membrane transporters, enhanced survival signaling, increased drug in activation as well as increased DNA repair processes compared to cancer cells. This allows CSCs to survive conventional therapy and thus contribute to chemoresistance for example. Adapted from Senthebane et al. [105].
Several other proteins associated with apoptosis are also involved in the survival of cancer cells and CSCs [106][107]. For example, several pro-survival proteins including BCL-2, B-cell lymphoma extra-large (Bcl-xL) and BCL-2-like-2 (BCL-W) have been found to be overexpressed in several cancer types including lymphoid cancer [108][109]. The overexpression of these pro-survival proteins has also been linked with carcinogenesis, with the blocking of these proteins and their associated pathways resulting in reduced tumor growth and enhanced response to chemotherapy [109][110][111].
Cytotoxic drugs target rapidly growing cancer cells, making them ineffective against slow dividing or dormant CSCs [21]. Viale and colleagues demonstrated that leukemia CSCs proliferate at a much lower rate than other cancer cells [112]. Therapies that target cancer cell cycling would therefore be ineffective against CSCs. Therapeutic agents such as paclitaxel would be unable to be less effective against slow dividing CSCs [113]. In addition, several studies demonstrate that CSCs show enhanced DNA damage repair capacity, with phosphorylation of repair enzymes observed in cancers such as breast and gliomas [114][115]. CSCs including glioma stem cells demonstrate great abilities at ROS scavenging thus protecting themselves against oxidative DNA damage [116][117][118]. Therapy itself has been shown to selectively increase CSCs in tumors. For example, Rizzo and colleagues demonstrated that CSCs are enriched in ovarian tumors after chemotherapy [119]. In addition, Levina and coworkers showed that chemotherapy can lead to the propagation of CSCs in lung cancer [120]. Thus, chemotherapy only targets the rapidly proliferating cancer cells leaving the CSCs to propagate the tumor after therapy. Chen and colleagues demonstrated that the drug temozolomide (TMZ) activates CSCs to produce cancer cells after therapy [121]. Qiu and colleagues demonstrated that elevated O6-methylguanine DNA methyltransferase (MGMT) expression and activity in glioma stem-like cells were responsible for temozolomide resistance [122]. Kurtova and coworkers also demonstrated that blockage of tumor repopulation by CSCs is effective at attenuating therapy resistance in bladder cancer [123]. Saito and colleagues demonstrated that inducing cell cycle re-entry through treatment with granulocyte colony-stimulating factor (G-CSF) allows normal chemotherapy to eliminate cancer cells effectively [124]. In addition, the induction of CSCs differentiation has been used successfully to increase CSCs sensitivity to commonly used cancer drugs. Lombardo and coworkers induced colorectal CSCs terminal differentiation via the use of bone morphogenic protein 4 (BMP4) and observed increased CSCs sensitization to standard chemotherapy [125]. Wang and coworkers used silibinin, which blocks colon CSCs self-renewal, resulting in reduced CSC population leading to reduced cancer cell proliferation [126]. Whilst several strategies have been developed to induce CSCs differentiation, all-trans retinoic acid (ATRA) is one of the common drugs used for this purpose [127][128].
2.2. Cancer Stem Cells and Angiogenesis
Many biological processes are dependent on the formation of new blood vessels, a process referred to as angiogenesis. Normal development and tissue repair and regeneration are especially dependent on new blood vessels for the supply of nutrients as well as the removal of toxic material [129][130]. Besides normal biological activities, angiogenesis is a requirement for tumor formation beyond a certain diameter [131][132]. During tumor formation, the usual delicate balance between pro-angiogenesis and anti-angiogenesis is altered, with pro-angiogenesis factors dominating [131]. New blood vessels sprout from pre-existing vessels within and around the tumor, fueling the rapid growth of the tumor [133][134]. The rapid growth of a tumor results in hypoxic conditions within the tumor. CSCs are known to release factors such as hypoxia-inducible factor 1 which induces the release of proangiogenic factors (Figure 4) [135][136]. Hypoxia has also been shown to fuel CSCs [137]. Hypoxia promotes CSC growth in several cancers via the upregulation of adaptive transcriptional programs, allowing CSCs to survive, invade and metastasize [137][138]. Soeda and colleagues demonstrated that hypoxia promotes the self-renewal capacity of CD133-positive human glioma-derived cancer stem cells [137]. Heddleston and colleagues showed that hypoxia promotes CSC self-renewal capabilities and stem-like phenotype even in non-stem cancer cell populations [139]. As reviewed by Heddleston and colleagues, CSC plasticity is influenced by hypoxia and new drugs must target such microenvironmental conditions for durable cancer treatment [140].
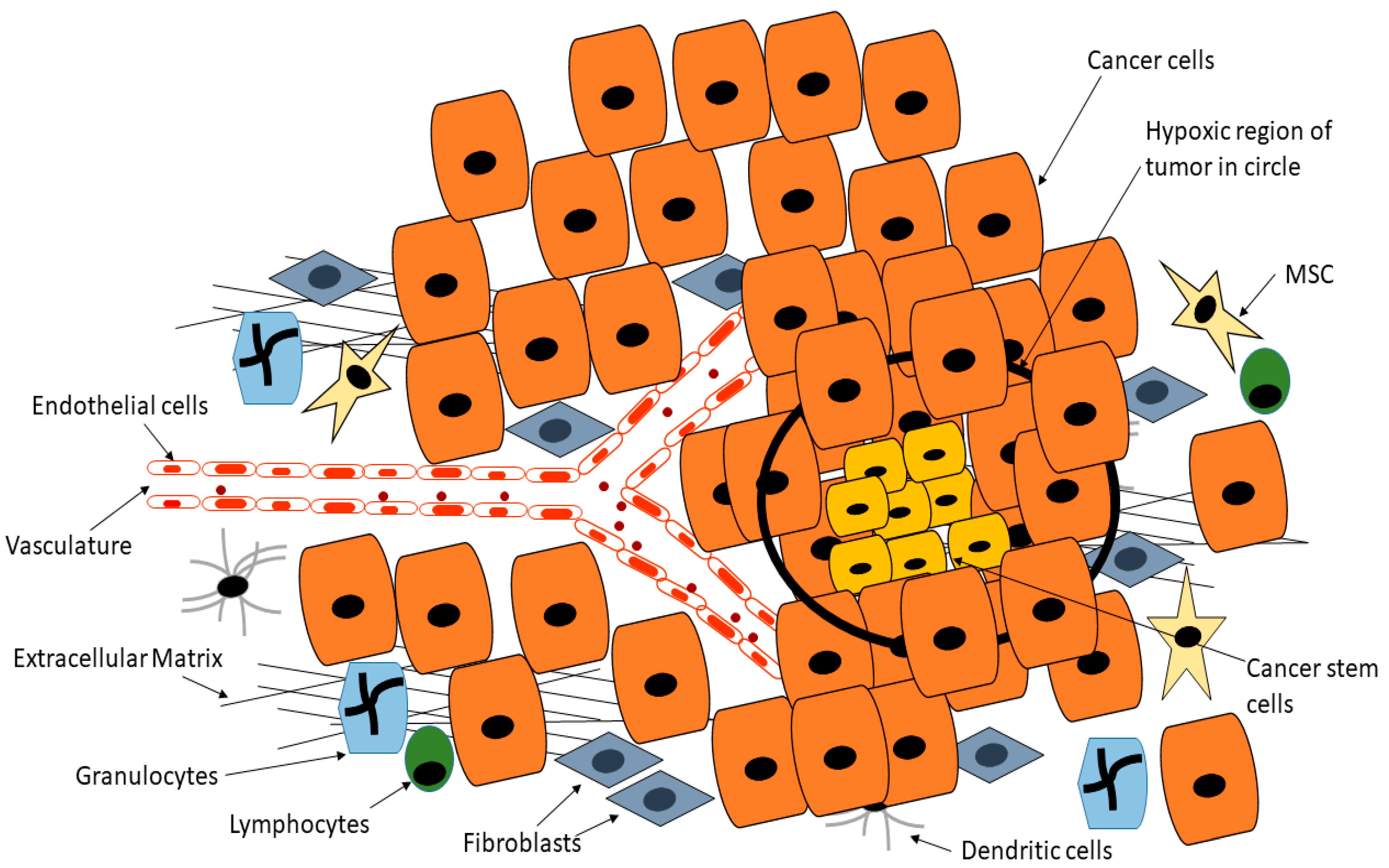
Figure 4. Cancer stem cells are able to reside deep within the tumor in hypoxic regions that are normally toxic to normal cells, whilst CSCs are able to release factors such as hypoxia-inducible factor 1 which induces the release of proangiogenic factors, this position means CSCs are inaccessible to drugs or are exposed to reduced drug doses. Adapted from Senthebane et al. [105].
Endothelial cells are also recruited to the tumor site. Endothelial cells express VEGFR and the binding of VEGF-A results in the activation of several signaling cascades involved in migration and ECM remodeling [136][141]. Survival pathways including the PI3K-Akt and the MEK-ERK cascades are activated and play key roles in the activation of endothelial cells to form new blood vessels [141]. Several cytokines are also known to be secreted by CSCs within the TME and these include IL-6 and TNF-α [142][143]. The secreted cytokines are involved in the recruitment of immune cells such as myeloid cells to further promote tumorigenesis [142][143]. Dysregulation of the Notch pathway has also been associated with tumor growth in general and survival of CSCs [144][145][146]. Several reports demonstrated that cells showing high expression of Notch signaling have elevated tumor-forming abilities and self-renewal capacity than those with less Notch activation [147][148][149]. Activation of the Notch pathway has also been associated with proangiogenic activity, with Notch ligand Jagged-1 promoting blood vessel formation [150][151][152].
In addition, matrix metalloproteases (MMPs) secreted by both cancer cells and stromal cells remodel the TME, allowing the creation of space for blood vessels formation as well as recruitment of different cells [153][154]. Due to the plasticity of CSCs, suggestions have been made to the effect that CSCs can give rise to endothelial cells and pericytes within and around the tumor [155][156]. Blood vessels within the TME are convoluted and “leaky”, resulting in fewer drugs able to reach cancer cells and CSCs deep within the TME. Tumor-derived cells are also able to intravasate and travel to distance sites, promoting metastasis in the process. Several studies have demonstrated the presence of circulating tumor-derived cells that are able to act as “seeds” for new tumors in distant sites [157][158]. Once the circulating cancer cells reach distant sites, they are able to extravasate and form new tumors in favorable microenvironments [10][159]. The formation of new tumors is dependent on CSCs successfully inducing angiogenesis to allow the exchange of nutrients and metabolic byproducts. Whilst it has been shown that stromal cells play a key role in inducing angiogenesis within tumors, CSCs are also involved in releasing angiogenic factors [160]. For example, Bao and colleagues demonstrated that glioma CSCs release VEGF resulting in increased microvascular density in malignant glioma [161]. Monzani and colleagues also showed that melanoma CSCs co-expressed CD133 and VEGF [162]. Maeda and colleagues also showed that pancreatic CSCs co-express CD133 and VEGF-C resulting in increased microvascular density [163]. It is also possible that cancer cells may enter a state of dormancy in which they remain until induced to proliferate and form new tumors [164][165].
2.3. Cancer Stem Cells and Epithelial to Mesenchymal Transition
Besides the influence of genetic and epigenetic mechanisms on the CSC phenotype, the TME within which CSCs are located plays a huge role in the CSC behavior [21]. As more data emerges the CSC field continues to change and be refined [110][166]. Overall, the CSC phenotype is dynamic and never constant. When CSCs undergo EMT, they acquire characteristics allowing them to migrate, invade surrounding tissues and metastasize [167]. EMT and CSC characteristics appear to share similar molecular pathways that are involved in invasion and migration of cancer cells from the primary tumor. In addition, transcriptional analysis of EMT and those associated with CSCs reveal significant overlap in gene expression including TGF-β, Hedgehog signaling and microRNAs [17]. EMT has been associated with poor prognosis in several cancers including esophageal and colon cancers [168][169]. Several signaling pathways have been identified to be key in modulating CSCs behavior including invasiveness and metastatic ability [170][171]. In addition, several markers identifying CSCs with invasive and metastatic abilities have been revealed including CD44v6 [172][173]. CD44 is specifically expressed by breast epithelial cells undergoing EMT [174]. EMT is characterized by the loss of cell to cell adhesion with cells becoming mesenchymal and markers such as E-cadherin lacking in such cells [175][176]. The loss of E-cadherin from the cell surface is accompanied by the expression of N-cadherin [177]. Histone deacetylation of the CDH1 promoter through the actions of DNMT and HDACs leads to gene silencing [178][179]. Histone methylation within the CDH1 promoter via the EZH2 and PRC2 complex is known to silence its expression [180].
EMT is influenced by several protein factors as well as microRNAs. For example, TGF-β has been regarded as a master regulator of EMT in certain cancers including breast and colorectal cancers [181]. Besides influencing cancer cells, TGF-β can also regulate CAFs with a net effect of promoting metastasis [182]. Furthermore, microRNA-200 family members have been shown to suppress EMT via binding to two transcription factors, zinc finger E-box-binding homeobox 1 (ZEB1) and ZEB2 [183][184]. Tellez and colleagues demonstrated that EMT can be induced by epigenetic mechanisms including chromatin remodeling through H3K27me3 enrichment as well as DNA methylation to sustain silencing of tumor-suppressive microRNAs, microRNA-200b, microRNA-200c and microRNA-205 [185]. Thus, silencing these microRNAs through tri-methylation of DNMT and H3K27 can induce EMT-like and CSC characteristics [185].
2.4. Cancer Stem Cells and Metabolic Activity
Recently, metabolic alterations have been identified to cause cells to acquire stem-cell-like characteristics [186]. These alterations and the subsequent acquisition of stem-cell-like characteristics are thought to be caused by epigenetic changes in adult stem cells as well as cancer cells. Based on the CSC theory, acquisition of stem-cell-like characteristics makes these cells achieve a higher status within the hierarchy through the expression of self-renewal and pluripotent genes [21][70]. According to Menendez and Alarcon, products of mutated metabolic enzymes can behave as oncometabolites, inducing epigenetic changes in genetic material and thus drive tumor initiation and progression [186]. This and more pieces of evidence point to the need for a full view of tumor initiation and progression and not just focus on cancer cells. Metabolic processes can thus be targeted to stop tumor initiation and progression. Specifically, the TME is characterized by low oxygen and glucose levels and thus tends to favor oxidative phosphorylation as the main supplier of energy [187]. Hypoxia has been shown to induce metabolic alterations resulting in acidosis in several cancers [188][189]. Lee and colleagues demonstrated that chemoresistance and enhanced oxidative phosphorylation are correlated [187]. Recent studies demonstrated that indeed, the targeting of oxidative phosphorylation has shown some success in inhibiting CSCs metabolic processes and proliferation in some cancers [190][191].
Inhibition of the mitochondrial complex III resulted in decreased breast CSCs [192]. When relapse occurs, CSCs have been shown to increase oxidative phosphorylation levels to pretreatment levels, demonstrating the importance of oxidative phosphorylation in chemoresistance [193]. The adipose tissue and adipose-derived cells are able to interact with CSCs and have been shown to promote fatty acid oxidation in CSCs and chemoresistance [194]. The mitochondria are also known to play a role in CSC chemoresistance [195]. This is unsurprising as the mitochondria are central to many cellular processes such as metabolism, signaling and apoptosis. Mitochondria have recently been shown to play key roles in CSC behavior [196]. Sancho and colleagues concluded that the removal of CSCs through targeting mitochondrial function might prevent cancer disease from recurring and thus prevent fatal disease [197]. In colon CSCs, tumorigenic ability was associated with enhanced mitochondrial functions [198]. Atovaquone has been used to inhibit the mitochondrial complex II resulting in decreased breast CSCs [192]. Isayev and colleagues demonstrated that inhibition of glucose metabolism through the use of 3-bromopyruvate inhibited pancreatic CSCs growth and resistance to gemcitabine [199]. Several other studies also showed that inhibition of mitochondrial function affect CSC proliferation and self-renewal capabilities [200][201].
2.5. Cancer Stem Cells and Epigenetic Reprogramming
A contributing factor to the complex intra- and inter-tumor heterogeneity and the resulting failure of many anticancer therapies comes from CSC epigenetic alterations. The heritable non-genetic changes to CSCs phenotypes are what are called epigenetic reprogramming of CSCs [202][203]. Most of the proteins and enzymes involved in epigenetic reprogramming of cells including histone modifications and DNA methylations have been well-characterized [204][205]. For example, histone methyltransferases (HMTs) are responsible for methylation of histones whilst histone acetyltransferases are responsible for the acetylation of histones [206]. Demethylation and deacetylation of histones are carried out by histone demethylases (HDMs) and histone deacetylases (HDACs) respectively [206]. When acetylated, histones are more loosely packed and can be accessed by RNA polymerases, allowing transcription of genes around a specific location. On the other hand, methylation can activate or repress gene transcription. For example, the acetylation of histone H3/H4 is linked to the transcription of genes [207][208]. In addition, H3 lysine 4 methylation is also linked to transcription of several genes [209][210]. In contrast, the methylation of H3 lysine 9 and 27 is linked to gene repression [211][212][213]. It has been observed that different patterns of histone modification produce variable transcriptional outcomes, with some giving rise to activation of genes and others to repression [214][215]. Various mechanisms are known to be involved in epigenetic gene regulation, from modifications of cytosines in DNA, covalent modifications of histones, the involvement of noncoding RNAs to chromatin remodeling [216][217][218].
CpG islands are regions of the genome containing a large number of CpG dinucleotide repeats and usually extend for 300–3000 base pairs [219]. In most cases, CpG islands are located close to gene promoters in humans [220]. DNMT in addition to histone modification determines whether transcription occurs or not. When CpG islands are unmethylated, transcription can take place. When CpG islands are methylated the chromatin becomes transcription-suppressive. Methylation of CpG islands is catalyzed by DNMT1, DNMT3A and DNMT3B. Several tumor suppressor genes are silenced via CpG island methylation [221]. Transcription can also be repressed via the Polycomb repressive complexes 1 and 2 (PRC1 and PRC2) [222][223]. Polycomb repressors are able to catalyze the trimethylation of histone 3 lysine 27 (H3K27me3) giving rise to repression of genes associated with many cellular processes such as differentiation, development and choice of lineage [222][224]. Collinson and colleagues demonstrated that Polycomb complex PRC2 mediates H3K27me3 via the histone methyltransferase EZH2, leading to transcriptional repression of several genes [225].
CSCs and their subsets display epigenetic alterations including histone modifications and this eventually contributes to the intratumor heterogeneity observed in many tumors [226]. Several epigenetic regulators have mutations leading to tumor formation and progression as a result of epigenetic dysregulation [227][228]. Several CSC markers including CD133 are known to be regulated by epigenetic alterations [229]. Tabu and colleagues demonstrated that the hypomethylation of the CD133 promoter influences its expression in gliomas [230]. Yi and colleagues observed abnormal DNA methylation of CD133, a CSC marker, in colorectal and glioblastoma tumors [229]. Gorodetska and colleagues observed that EZH2/BRCA1 signaling mechanisms play an important role in the maintenance of prostate CSCs properties [231]. EMT aid in the generation of cells with stem cell characteristics and is modulated by epigenetic mechanisms [232][233]. The involvement of epigenetic mechanisms from CSC formation to maintenance makes epigenetics a therapeutic target in CSCs. Small compound inhibitors with the ability to induce differentiation in CSCs are therefore promising drugs targeting this population of tumor cells.
Several signaling pathways are crucial in facilitating the growth of CSCs and the maintenance of the CSC phenotype. Such signaling pathways include Hedgehog, Notch, JAK-STAT and Wnt-β- catenin signaling [234][235]. It is important to note that these same pathways are also important in regulating self-renewal in normal stem cells [236][237]. Several mutations have been observed in genes along these pathways in many human cancers. Signaling pathways such as Wnt and Notch have been observed in breast cancers for example [238] and in vitro work demonstrated that the overexpression of these pathways is associated with tumorigenicity [148][149]. Triple-negative breast cancer cells demonstrate increased Notch signaling and Notch signaling is associated with CD44 expression in colon cancer cells [151][239]. On the other hand, Wnt-β-catenin signaling has been observed to be associated with cancer stemness and heterogeneity [240]. Several members of the Wnt-β–catenin pathway have been linked to the induction of EMT in several cancers [241]. The hedgehog signaling pathway has been associated with self-renewal in many cancers including breast cancer and gliomas [242][243]. The hedgehog pathway has also been associated with EMT and invasion and migration [244][245].
Most of the above-mentioned signaling pathways are modulated by epigenetic mechanisms [246]. Under normal conditions, most of these pathways are involved in the propagation of CSCs, maintenance of the CSC phenotype as well as in embryonic development [247]. Several regulators of the above-mentioned pathways have been shown to have epigenetic alterations in CSCs. For example, decreased acetylation of H3K16 as well as enhanced H3K27 trimethylation is associated with DKK1 promoter silencing [248]. High levels of histone acetylation are observed at the promoter region of Notch receptor–ligand JAGGED2, resulting in Notch signaling activation in multiple myeloma cells [249]. In colorectal cancer, two Notch signaling targets, HES1 and HES2, show decreased promoter H3K27 methylation, resulting in gene activation [250][251][252]. Rhabdoid tumors show decreased or inactivation of SNF5, a member of chromatin remodeler complex SWI/SNF, leading to activation of Hedgehog signaling [253][254]. Furthermore, the activation of Gli1 and Gli2, downstream effectors of the Hedgehog signaling pathway, require HDAC1 [255][256][257]. As a result of the integration of genetic, epigenetic mechanisms and other factors, CSCs survival and maintenance are promoted.
The KMT2/MLL gene is known to encode for an HMT that influences many cellular processes [258][259]. MLL fusion proteins are present in several CSCs and have been shown to be involved in carcinogenesis in several cancers [260][261]. For example, Krivtsov and colleagues demonstrated that leukemia stem cells, with the MLL-AF9 fusion protein, can maintain the identity of progenitors from which they arose while at the same time activating stem-cell- or self-renewal-associated program [262]. Somervaille and colleagues also demonstrated that the hierarchical maintenance of MLL-myeloid leukemia stem cells utilizes a transcriptional program involving transcription/chromatin regulatory factors Myb, Hmgb3 and Cbx5 [263]. Several mutations have also been identified in histone-encoding genes. Lewis and colleagues showed that the blockage of PRC2 activity via the gain-of-function H3 mutation was prevalent in pediatric glioblastoma [264]. Furthermore, several DNMTs are mutated in acute myeloid leukemia and have been suggested to result in the formation of leukemia stem cells [265][266][267].
CSCs have been shown to play important roles in the propagation, growth and metastasis of colorectal cancer (CRC). Several genetic and epigenetic changes have been observed in CSCs in CRC. For example, the hypermethylation of several tumor suppressor gene promoters including p16, retinoblastoma, SFRP and MLH1, has been widely reported in many studies [268][269][270]. One of the driver mutations in CRC is the APC mutation, which influences the activities of DNMTs [271]. Increased levels of DNMT1 are thought to suppress the transcription of APC, a tumor suppressor gene in CRC [272][273]. The levels of DNMT1 in CSCs have been shown to be involved in CRC initiation and progression, directly linking epigenetic mechanisms to CSC-directed tumorigenesis [274]. Pathania and colleagues showed that DNMT1 is important for mammary and CSC maintenance and tumorigenesis [275].
References
- Graham, S.M.; Jorgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002, 99, 319–325.
- Holyoake, T.L.; Jiang, X.; Jorgensen, H.G.; Graham, S.; Alcorn, M.J.; Laird, C.; Eaves, A.C.; Eaves, C.J. Primitive quiescent leukemic cells from patients with chronic myeloid leukemia spontaneously initiate factor-independent growth in vitro in association with up-regulation of expression of interleukin-3. Blood 2001, 97, 720–728.
- Paget, S. The Distribution of Secondary Growths in Cancer of the Breast. Lancet 1889, 133, 571–573.
- Oskarsson, T.; Batlle, E.; Massagué, J. Metastatic stem cells: Sources, niches, and vital pathways. Cell Stem Cell 2014, 14, 306–321.
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306.
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775.
- Leon, G.; MacDonagh, L.; Finn, S.P.; Cuffe, S.; Barr, M.P. Cancer stem cells in drug resistant lung cancer: Targeting cell surface markers and signaling pathways. Pharmacol. Ther. 2016, 158, 71–90.
- Maugeri-Saccà, M.; Bartucci, M.; De Maria, R. DNA Damage Repair Pathways in Cancer Stem Cells. Mol. Cancer Ther. 2012, 11, 1627–1636.
- Maugeri-Saccà, M.; Vigneri, P.; De Maria, R. Cancer stem cells and chemosensitivity. Clin. Cancer Res. 2011, 17, 4942–4947.
- Turdo, A.; Veschi, V.; Gaggianesi, M.; Chinnici, A.; Bianca, P.; Todaro, M.; Stassi, G. Meeting the Challenge of Targeting Cancer Stem Cells. Front. Cell Dev. Boil. 2019, 7, 16.
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737.
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648.
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988.
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Wicinski, J.; Cervera, N.; Finetti, P.; Hur, M.-H.; Diebel, M.E.; Monville, F.; Dutcher, J.; et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009, 69, 1302–1313.
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2007, 15, 504–514.
- Agliano, A.; Calvo, A.; Box, C. The challenge of targeting cancer stem cells to halt metastasis. Semin. Cancer Boil. 2017, 44, 25–42.
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143.
- Aartsma-Rus, A.; Van Putten, M. The use of genetically humanized animal models for personalized medicine approaches. Dis. Model. Mech. 2019, 13, dmm041673.
- Agarwal, Y.; Beatty, C.; Biradar, S.; Castronova, I.; Ho, S.; Melody, K.; Bility, M.T. Moving beyond the mousetrap: Current and emerging humanized mouse and rat models for investigating prevention and cure strategies against HIV infection and associated pathologies. Retrovirology 2020, 17, 1–11.
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458.
- Dzobo, K.; Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Mwapagha, L.M.; Al-Awwad, N.; Dandara, C.; Parker, M.I. Cancer Stem Cell Hypothesis for Therapeutic Innovation in Clinical Oncology? Taking the Root Out, Not Chopping the Leaf. OMICS A J. Integr. Boil. 2016, 20, 681–691.
- Bozorgi, A.; Khazaei, M.; Khazaei, M.R. New Findings on Breast Cancer Stem Cells: A Review. J. Breast Cancer 2015, 18, 303–312.
- Singh, S.R. Gastric cancer stem cells: A novel therapeutic target. Cancer Lett. 2013, 338, 110–119.
- MacDonagh, L.; Gray, S.G.; Breen, E.; Cuffe, S.; Finn, S.P.; O’Byrne, K.J.; Barr, M.P. Lung cancer stem cells: The root of resistance. Cancer Lett. 2016, 372, 147–156.
- Dzobo, K.; Rowe, A.; Senthebane, D.A.; Almazyadi, M.A.; Patten, V.; Parker, M.I. Three-Dimensional Organoids in Cancer Research: The Search for the Holy Grail of Preclinical Cancer Modeling. OMICS J. Integr. Boil. 2018, 22, 733–748.
- Ortiz-Sánchez, E.; Santiago-López, L.; Cruz-Domínguez, V.B.; Toledo-Guzmán, M.E.; Hernández-Cueto, D.; Muñiz-Hernández, S.; Garrido, E.; De León, D.C.; García-Carrancá, A. Characterization of cervical cancer stem cell-like cells: Phenotyping, stemness, and human papilloma virus co-receptor expression. Oncotarget 2016, 7, 31943–31954.
- Hou, T.; Zhang, W.; Tong, C.; Kazobinka, G.; Huang, X.; Huang, Y.; Zhang, Y. Putative stem cell markers in cervical squamous cell carcinoma are correlated with poor clinical outcome. BMC Cancer 2015, 15, 785.
- Tyagi, A.; Vishnoi, K.; Mahata, S.; Verma, G.; Srivastava, Y.; Masaldan, S.; Roy, B.G.; Bharti, A.C.; Das, B.C. Cervical Cancer Stem Cells Selectively Overexpress HPV Oncoprotein E6 that Controls Stemness and Self-Renewal through Upregulation of HES1. Clin. Cancer Res. 2016, 22, 4170–4184.
- Ming, X.-Y.; Fu, L.; Zhang, L.-Y.; Qin, Y.-R.; Cao, T.-T.; Chan, K.W.; Ma, S.K.Y.; Xie, D.; Guan, X.-Y. Integrin α7 is a functional cancer stem cell surface marker in oesophageal squamous cell carcinoma. Nat. Commun. 2016, 7, 13568.
- Zhao, J.-S.; Li, W.-J.; Ge, D.; Zhang, P.-J.; Li, J.-J.; Lu, C.-L.; Ji, X.-D.; Guan, D.-X.; Gao, H.; Xu, L.-Y.; et al. Tumor Initiating Cells in Esophageal Squamous Cell Carcinomas Express High Levels of CD44. PLoS ONE 2011, 6, e21419.
- Yuan, Z.-X.; Mo, J.; Zhao, G.; Shu, G.; Fu, H.-L.; Zhao, W. Targeting Strategies for Renal Cell Carcinoma: From Renal Cancer Cells to Renal Cancer Stem Cells. Front. Pharmacol. 2016, 7, 423.
- Peired, A.; Sisti, A.; Romagnani, P. Renal Cancer Stem Cells: Characterization and Targeted Therapies. Stem Cells Int. 2016, 2016, 1–12.
- Cheng, B.; Yang, G.; Jiang, R.; Cheng, Y.; Yang, H.; Pei, L.; Qiu, X. Cancer stem cell markers predict a poor prognosis in renal cell carcinoma: A meta-analysis. Oncotarget 2016, 7, 65862–65875.
- Fan, F.; Bellister, S.; Lü, J.; Ye, X.; Boulbès, D.R.; Tozzi, F.; Sceusi, E.; Kopetz, S.; Tian, F.; Xia, L.; et al. The requirement for freshly isolated human colorectal cancer (CRC) cells in isolating CRC stem cells. Br. J. Cancer 2014, 112, 539–546.
- Dalerba, P.; Kalisky, T.; Sahoo, D.; Rajendran, P.S.; Rothenberg, M.E.; Leyrat, A.A.; Sim, S.; Okamoto, J.; Johnston, D.M.; Qian, D.; et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat. Biotechnol. 2011, 29, 1120–1127.
- Yeung, T.M.; Gandhi, S.C.; Wilding, J.L.; Muschel, R.; Bodmer, W.F.B.J. Cancer stem cells from colorectal cancer-derived cell lines. Proc. Nat. Acad. Sci. USA 2010, 107, 3722–3727.
- Kemper, K.; Prasetyanti, P.R.; De Lau, W.; Rodermond, H.; Clevers, H.; Medema, J.P. Monoclonal Antibodies Against Lgr5 Identify Human Colorectal Cancer Stem Cells. Stem Cells 2012, 30, 2378–2386.
- Yang, Z.F.; Ho, D.W.; Ng, M.N.; Lau, C.K.; Yu, W.C.; Ngai, P.; Chu, P.W.; Lam, C.T.; Poon, R.T.; Fan, S.T. Significance of CD90+ Cancer Stem Cells in Human Liver Cancer. Cancer Cell 2008, 13, 153–166.
- Kimura, O.; Takahashi, T.; Ishii, N.; Inoue, Y.; Ueno, Y.; Kogure, T.; Fukushima, K.; Shiina, M.; Yamagiwa, Y.; Kondo, Y.; et al. Characterization of the epithelial cell adhesion molecule (EpCAM) + cell population in hepatocellular carcinoma cell lines. Cancer Sci. 2010, 101, 2145–2155.
- Al Faraj, A.; Shaik, A.S.; Al Sayed, B.; Halwani, R.; Al Jammaz, I. Specific targeting and noninvasive imaging of breast cancer stem cells using single-walled carbon nanotubes as novel multimodality nanoprobes. Nanomedicine 2016, 11, 31–46.
- Nie, S.; McDermott, S.P.; Deol, Y.; Tan, Z.; Wicha, M.S.; Lubman, D.M. A quantitative proteomics analysis of MCF7 breast cancer stem and progenitor cell populations. Proteomics. 2015, 15, 3772–3783.
- A Theodoropoulos, P.; Chiotaki, R.; Polioudaki, H. Stem cell technology in breast cancer: Current status and potential applications. Stem Cells Cloning Adv. Appl. 2016, 9, 17–29.
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009, 27, 1006–1020.
- Zhang, C.; Li, C.; He, F.; Cai, Y.; Yang, H. Identification of CD44+CD24+ gastric cancer stem cells. J. Cancer Res. Clin. Oncol. 2011, 137, 1679–1686.
- Mao, J.; Fan, S.; Ma, W.; Fan, P.; Wang, B.; Zhang, J.; Wang, H.; Tang, B.; Zhang, Q.; Yu, X.; et al. Roles of Wnt/β-catenin signaling in the gastric cancer stem cells proliferation and salinomycin treatment. Cell Death Dis. 2014, 5, e1039.
- Beier, D.; Hau, P.; Proescholdt, M.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. CD133+ and CD133− glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007, 67, 4010–4015.
- Yan, X.; Ma, L.; Yi, D.; Yoon, J.-G.; Diercks, A.; Foltz, G.; Price, N.D.; Hood, L.E.; Tian, Q. A CD133-related gene expression signature identifies an aggressive glioblastoma subtype with excessive mutations. Proc. Nat. Acad. Sci. USA 2011, 108, 1591–1596.
- Wang, J.C.Y.; Dick, J.E. Cancer stem cells: Lessons from leukemia. Trends Cell Boil. 2005, 15, 494–501.
- Afify, S.M.M.; Hassan, G.; Osman, A.; Calle, A.S.; Nawara, H.M.; Zahra, M.H.; El-Ghlban, S.; Mansour, H.; Alam, J.; Abu Quora, H.A.; et al. Metastasis of Cancer Stem Cells Developed in the Microenvironment of Hepatocellular Carcinoma. Bioengineering 2019, 6, 73.
- Hope, K.J.; Jin, L.; Dick, J.E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol. 2004, 5, 738–743.
- Inoue, A.; Kobayashi, C.I.; Shinohara, H.; Miyamoto, K.; Yamauchi, N.; Yuda, J.; Akao, Y.; Minami, Y. Chronic myeloid leukemia stem cells and molecular target therapies for overcoming resistance and disease persistence. Int. J. Hematol. 2018, 108, 365–370.
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Investig. 2011, 121, 396–409.
- Foster, R.; Buckanovich, R.J.; Rueda, B.R. Ovarian cancer stem cells: Working towards the root of stemness. Cancer Lett. 2013, 338, 147–157.
- Burgos-Ojeda, D.; Rueda, B.R.; Buckanovich, R.J. Ovarian cancer stem cell markers: Prognostic and therapeutic implications. Cancer Lett. 2012, 322, 1–7.
- Collins, A.T.; Maitland, N.J. Prostate cancer stem cells. Eur. J. Cancer 2006, 42, 1213–1218.
- Chen, X.; Li, Q.; Liu, X.; Liu, C.; Liu, R.; Rycaj, K.; Zhang, D.; Liu, B.; Jeter, C.; Calhoun-Davis, T.; et al. Defining a Population of Stem-like Human Prostate Cancer Cells That Can Generate and Propagate Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 4505–4516.
- Liu, X.; Chen, X.; Rycaj, K.; Chao, H.-P.; Deng, Q.; Jeter, C.R.; Liu, C.; Honorio, S.; Li, H.; Davis, T.; et al. Systematic dissection of phenotypic, functional, and tumorigenic heterogeneity of human prostate cancer cells. Oncotarget 2015, 6, 23959–23986.
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323.
- Lee, C.J.; Dosch, J.; Simeone, D.M. Pancreatic Cancer Stem Cells. J. Clin. Oncol. 2008, 26, 2806–2812.
- Li, C.; Lee, C.J.; Simeone, D.M. Identification of Human Pancreatic Cancer Stem Cells. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2009; Volume 568, pp. 161–173.
- La Porta, C.A.M. Cancer Stem Cells: Lessons from Melanoma. Stem Cell Rev. Rep. 2008, 5, 61–65.
- Kumar, D.; Gorain, M.; Kundu, G.; Kundu, G.C. Therapeutic implications of cellular and molecular biology of cancer stem cells in melanoma. Mol. Cancer 2017, 16, 7.
- Chinn, S.B.; Darr, O.A.; Owen, J.H.; Bellile, E.; McHugh, J.B.; Spector, M.E.; Papagerakis, S.M.; Chepeha, U.B.; Bradford, C.R.; Carey, T.E.; et al. Cancer stem cells: Mediators of tumorigenesis and metastasis in head and neck squamous cell carcinoma. Head Neck 2014, 37, 317–326.
- Kim, J.; Shin, J.H.; Chen, C.-H.; Cruz, L.; Farnebo, L.; Yang, J.; Borges, P.; Kang, G.; Mochly-Rosen, D.; Sunwoo, J.B. Targeting aldehyde dehydrogenase activity in head and neck squamous cell carcinoma with a novel small molecule inhibitor. Oncotarget 2017, 8, 52345–52356.
- Lan, J.; Huang, B.; Liu, R.; Ju, X.; Zhou, Y.; Jiang, J.; Liang, W.; Shen, Y.; Li, F.; Pang, L. Expression of cancer stem cell markers and their correlation with pathogenesis in vascular tumors. Int. J. Clin. Exp. Pathol. 2015, 8, 12621–12633.
- Veselska, R.; Skoda, J.; Neradil, J. Detection of cancer stem cell markers in sarcomas. Klin. Onkol. 2012, 25, 16–20.
- Visvader, J.E.; Lindeman, G.J. Cancer Stem Cells: Current Status and Evolving Complexities. Cell Stem Cell 2012, 10, 717–728.
- Miyoshi, N.; Mizushima, T.; Doki, Y.; Mori, M. Cancer stem cells in relation to treatment. Jpn. J. Clin. Oncol. 2018, 49, 232–237.
- Toh, T.B.; Lim, J.J.; Chow, E.K.-H. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 29.
- Dawood, S.; Austin, L.; Cristofanilli, M. Cancer stem cells: Implications for cancer therapy. Oncology (Williston Park. N. Y.) 2014, 28, 1101–1107.
- Steinbichler, T.B.; Dudas, J.; Skvortsov, S.; Ganswindt, U.; Riechelmann, H.; Skvortsova, I.-I. Therapy resistance mediated by cancer stem cells. Semin. Cancer Boil. 2018, 53, 156–167.
- Dymock, B.W. The rise of epigenetic drug discovery. Futur. Med. Chem. 2016, 8, 1523–1524.
- Hsu, L.C.; Chang, W.C.; Hoffmann, I.; Duester, G. Molecular analysis of two closely related mouse aldehyde dehydrogenase genes: Identification of a role for Aldh1, but not Aldh-pb, in the biosynthesis of retinoic acid. Biochem. J. 1999, 339, 387–395.
- Yoshida, A.; Rzhetsky, A.; Hsu, L.C.; Chang, C. Human aldehyde dehydrogenase gene family. JBIC J. Boil. Inorg. Chem. 1998, 251, 549–557.
- Eirew, P.; Kannan, N.; Knapp, D.J.H.F.; Vailllant, F.; Emerman, J.T.; Lindeman, G.J.; Visvader, J.E.; Eaves, C.J. Aldehyde Dehydrogenase Activity Is a Biomarker of Primitive Normal Human Mammary Luminal Cells. Stem Cells 2012, 30, 344–348.
- Ioannou, M.; Serafimidis, I.; Arnes, L.; Sussel, L.; Singh, S.; Vasiliou, V.; Gavalas, A. ALDH1B1 is a potential stem/progenitor marker for multiple pancreas progenitor pools. Dev. Boil. 2012, 374, 153–163.
- Singh, S.; Brocker, C.; Koppaka, V.; Chen, Y.; Jackson, B.C.; Matsumoto, A.; Thompson, D.C.; Vasiliou, V. Aldehyde Dehydrogenases in Cellular Responses to Oxidative/electrophilic Stress. Free Radic. Boil. Med. 2012, 56, 89–101.
- Vassalli, G. Aldehyde Dehydrogenases: Not Just Markers, but Functional Regulators of Stem Cells. Stem Cells Int. 2019, 2019, 1–15.
- Ciccone, V.; Terzuoli, E.; Donnini, S.; Giachetti, A.; Morbidelli, L.; Ziche, M. Stemness marker ALDH1A1 promotes tumor angiogenesis via retinoic acid/HIF-1α/VEGF signalling in MCF-7 breast cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 311.
- Vogler, T.; Kriegl, L.; Horst, D.; Engel, J.; Sagebiel, S.; Schäffauer, A.J.; Kirchner, T.; Jung, A. The expression pattern of aldehyde dehydrogenase 1 (ALDH1) is an independent prognostic marker for low survival in colorectal tumors. Exp. Mol. Pathol. 2012, 92, 111–117.
- Hoogen, C.V.D.; Van Der Horst, G.; Cheung, H.; Buijs, J.T.; Lippitt, J.M.; Hamdy, F.C.; Eaton, C.L.; Thalmann, G.N.; Cecchini, M.G.; Pelger, R.C.; et al. High Aldehyde Dehydrogenase Activity Identifies Tumor-Initiating and Metastasis-Initiating Cells in Human Prostate Cancer. Cancer Res. 2010, 70, 5163–5173.
- Ueda, K.; Ogasawara, S.; Akiba, J.; Nakayama, M.; Todoroki, K.; Ueda, K.; Sanada, S.; Suekane, S.; Noguchi, M.; Matsuoka, K.; et al. Aldehyde Dehydrogenase 1 Identifies Cells with Cancer Stem Cell-Like Properties in a Human Renal Cell Carcinoma Cell Line. PLoS ONE 2013, 8, e75463.
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555–567.
- Ma, S.K.Y.; Lee, T.K.; Zheng, B.-J.; Chan, K.W.; Guan, X.-Y.; Lee, T.K.W. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2007, 27, 1749–1758.
- Jin, X.; Zhao, Y.; Qian, J.; Tang, J.; Zhan, X.-D. Aldehyde dehydrogenase 1 can be used as a new marker of cancer stem cells in laryngeal cancer cells in vitro. Zhonghua Zhong Liu Za Zhi (Chin. J. Oncol.) 2011, 33, 900–904.
- Afify, S.M.M.; Seno, M. Conversion of Stem Cells to Cancer Stem Cells: Undercurrent of Cancer Initiation. Cancers 2019, 11, 345.
- Tanei, T.; Morimoto, K.; Shimazu, K.; Kim, S.J.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of Breast Cancer Stem Cells Identified by Aldehyde Dehydrogenase 1 Expression with Resistance to Sequential Paclitaxel and Epirubicin-Based Chemotherapy for Breast Cancers. Clin. Cancer Res. 2009, 15, 4234–4241.
- Morimoto, K.; Kim, S.J.; Tanei, T.; Shimazu, K.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Terada, N.; Noguchi, S. Stem cell marker aldehyde dehydrogenase 1-positive breast cancers are characterized by negative estrogen receptor, positive human epidermal growth factor receptor type 2, and high Ki67 expression. Cancer Sci. 2009, 100, 1062–1068.
- Rausch, V.; Liu, L.; Kallifatidis, G.; Baumann, B.; Mattern, J.; Gladkich, J.; Wirth, T.; Schemmer, P.; Buchler, M.W.; Zöller, M.; et al. Synergistic Activity of Sorafenib and Sulforaphane Abolishes Pancreatic Cancer Stem Cell Characteristics. Cancer Res. 2010, 70, 5004–5013.
- Yip, N.C.; Fombon, I.S.; Liu, P.; Brown, S.; Kannappan, V.; Armesilla, A.L.; Xu, B.; Cassidy, J.; Darling, J.L.; Wang, W. Disulfiram modulated ROS–MAPK and NFκB pathways and targeted breast cancer cells with cancer stem cell-like properties. Br. J. Cancer 2011, 104, 1564–1574.
- Locher, K.P. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Boil. 2016, 23, 487–493.
- Begicevic, R.-R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362.
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284.
- Lario, A.P.; García, C.B.; Elizondo, M.E.; Lobo, C. Expression of proteins associated with multidrug resistance and resistance to chemotherapy in lung cancer. Arch. Bronconeumol. (Engl. Ed.) 2007, 43, 479–484.
- Leonessa, F.; Clarke, R. ATP binding cassette transporters and drug resistance in breast cancer. Endocr. Relat. Cancer 2003, 10, 43–73.
- Boesch, M.; Zeimet, A.G.; Rumpold, H.; Gastl, G.; Sopper, S.; Wolf, D. Drug Transporter-Mediated Protection of Cancer Stem Cells from Ionophore Antibiotics. Stem Cells Transl. Med. 2015, 4, 1028–1032.
- Xiong, B.; Ma, L.; Hu, X.; Zhang, C.; Cheng, Y. Characterization of side population cells isolated from the colon cancer cell line SW480. Int. J. Oncol. 2014, 45, 1175–1183.
- Wright, M.H.; Calcagno, A.M.; Salcido, C.D.; Carlson, M.D.; Ambudkar, S.V.; Varticovski, L. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008, 10, R10.
- Frank, M.H.; Margaryan, A.; Sadée, W.; Huang, Y.; Schatton, T.; Waaga-Gasser, A.M.; Waaga-Gasser, A.M.; Sayegh, M.H. ABCB5-Mediated Doxorubicin Transport and Chemoresistance in Human Malignant Melanoma. Cancer Res. 2005, 65, 4320–4333.
- Shi, G.-M.; Xu, Y.; Fan, J.; Zhou, J.; Yang, X.-R.; Qiu, S.-J.; Liao, Y.; Wu, W.-Z.; Ji, Y.; Ke, A.-W.; et al. Identification of side population cells in human hepatocellular carcinoma cell lines with stepwise metastatic potentials. J. Cancer Res. Clin. Oncol. 2008, 134, 1155–1163.
- Moitra, K. Overcoming Multidrug Resistance in Cancer Stem Cells. BioMed Res. Int. 2015, 2015, 1–8.
- Lou, H.; Dean, M. Targeted therapy for cancer stem cells: The patched pathway and ABC transporters. Oncogene 2007, 26, 1357–1360.
- Marcelletti, J.F.; Multani, P.S.; Lancet, J.E.; Baer, M.R.; Sikic, B.I. Leukemic blast and natural killer cell P-glycoprotein function and inhibition in a clinical trial of zosuquidar infusion in acute myeloid leukemia. Leuk. Res. 2009, 33, 769–774.
- Tang, R.; Faussat, A.-M.; Perrot, J.-Y.; Marjanovic, Z.; Cohen, S.; Storme, T.; Morjani, H.; Legrand, O.; Marie, J.-P. Zosuquidar restores drug sensitivity in P-glycoprotein expressing acute myeloid leukemia (AML). BMC Cancer 2008, 8, 51.
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Al Mazeedi, M.A.M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586.
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Sun, H.-R.; Wang, S.; Yan, S.-C.; Zhang, Y.; Nelson, P.J.; Jia, H.-L.; Qin, L.-X.; Dong, Q.-Z. Therapeutic Strategies Targeting Cancer Stem Cells and Their Microenvironment. Front. Oncol. 2019, 9, 9.
- Strasser, A.; Harris, A.W.; Bath, M.L.; Cory, S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 1990, 348, 331–333.
- Todaro, M.; Alea, M.P.; Di Stefano, A.B.; Cammareri, P.; Vermeulen, L.; Iovino, F.; Tripodo, C.; Russo, A.; Gulotta, G.; Medema, J.P.; et al. Colon Cancer Stem Cells Dictate Tumor Growth and Resist Cell Death by Production of Interleukin-4. Cell Stem Cell 2007, 1, 389–402.
- Sun, Q.; Wang, Y.; Desgrosellier, J.S. Combined Bcl-2/Src inhibition synergize to deplete stem-like breast cancer cells. Cancer let. 2019, 457, 40–46.
- Viale, A.; De Franco, F.; Orleth, A.; Cambiaghi, V.; Giuliani, V.; Bossi, D.; Ronchini, C.; Ronzoni, S.; Muradore, I.; Monestiroli, S.; et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature 2009, 457, 51–56.
- Gascoigne, K.E.; Taylor, S.S.; Sridharan, D.; Brown, M.; Lambert, W.C.; McMahon, L.W. How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 2009, 122, 2579–2585.
- Mccord, A.M.; Jamal, M.; Williams, E.S.; Camphausen, K.; Tofilon, P.J. CD133+ glioblastoma stem-like cells are radiosensitive with a defective DNA damage response compared with established cell lines. Clin. Cancer Res. 2009, 15, 5145–5153.
- Gallmeier, E.; Hermann, P.C.; Mueller, M.-T.; Machado, J.G.; Ziesch, A.; De Toni, E.N.; Palagyi, A.; Eisen, C.; Ellwart, J.W.; Rivera, J.; et al. Inhibition of Ataxia Telangiectasia- and Rad3 -Related Function Abrogates the In Vitro and In Vivo Tumorigenicity of Human Colon Cancer Cells Through Depletion of the CD133+ Tumor-Initiating Cell Fraction. Stem Cells 2011, 29, 418–429.
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760.
- Cohen, A.A.; Geva-Zatorsky, N.; Eden, E.; Morgenstern, M.F.; Issaeva, I.; Sigal, A.; Milo, R.; Cohen-Saidon, C.; Liron, Y.; Kam, Z.; et al. Dynamic Proteomics of Individual Cancer Cells in Response to a Drug. Science 2008, 322, 1511–1516.
- Raha, D.; Wilson, T.R.; Peng, J.; Peterson, D.; Yue, P.; Evangelista, M.; Wilson, C.; Merchant, M.; Settleman, J. The Cancer Stem Cell Marker Aldehyde Dehydrogenase Is Required to Maintain a Drug-Tolerant Tumor Cell Subpopulation. Cancer Res. 2014, 74, 3579–3590.
- Rizzo, S.; Hersey, J.M.; Mellor, P.; Dai, W.; Santos-Silva, A.; Liber, D.; Luk, L.; Titley, I.; Carden, C.P.; Box, G.; et al. Ovarian cancer stem cell like side populations are enriched following chemotherapy and overexpress EZH2. Mol. Cancer Ther. 2011, 10, 325–335.
- Levina, V.; Marrangoni, A.M.; Demarco, R.; Gorelik, E.; Lokshin, A.E. Drug-Selected Human Lung Cancer Stem Cells: Cytokine Network, Tumorigenic and Metastatic Properties. PLoS ONE 2008, 3, e3077.
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526.
- Qiu, Z.-K.; Shen, N.; Chen, Y.-S.; Yang, Q.-Y.; Guo, C.-C.; Feng, B.-H.; Chen, Z.-P. Enhanced MGMT expression contributes to temozolomide resistance in glioma stem-like cells. Chin. J. Cancer 2014, 33, 115–122.
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.; Krasnow, R.E.; Lerner, S.P.; Chen, F.; Roh, T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2014, 517, 209–213.
- Saito, Y.; Uchida, N.; Tanaka, S.; Suzuki, N.; Tomizawa-Murasawa, M.; Sone, A.; Najima, Y.; Takagi, S.; Aoki, Y.; Wake, A.; et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat. Biotechnol. 2010, 28, 275–280.
- Lombardo, Y.; Scopelliti, A.; Cammareri, P.; Todaro, M.; Iovino, F.; Vitiani, L.R.; Gulotta, G.; Dieli, F.; De Maria, R.; Stassi, G. Bone Morphogenetic Protein 4 Induces Differentiation of Colorectal Cancer Stem Cells and Increases Their Response to Chemotherapy in Mice. Gastroenterology 2011, 140, 297–309.e6.
- Wang, J.-Y.; Chang, C.-C.; Chiang, C.-C.; Chen, W.-M.; Hung, S.-C. Silibinin suppresses the maintenance of colorectal cancer stem-like cells by inhibiting PP2A/AKT/mTOR pathways. J. Cell. Biochem. 2011, 113, 1733–1743.
- Nowak, D.; Stewart, D.; Koeffler, H.P. Differentiation therapy of leukemia: 3 decades of development. Blood 2009, 113, 3655–3665.
- Zhou, G.; Zhang, J.; Wang, Z.-Y.; Chen, S.-J.; Chen, Z. Treatment of acute promyelocytic leukaemia with all-trans retinoic acid and arsenic trioxide: A paradigm of synergistic molecular targeting therapy. Philos. Trans. R Soc. B Boil. Sci. 2007, 362, 959–971.
- Hanahan, D.; Folkman, J. Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell 1996, 86, 353–364.
- Pour, L.; Hájek, R.; Buchler, T.; Maisnar, V.; Smolej, L. Angiogenesis and antiangiogenic cancer therapy. Vnitrni Lek. 2004, 50, 930–938.
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18.
- Pandya, N.; Dhalla, N.S.; Santani, D.D. Angiogenesis—A new target for future therapy. Vasc. Pharmacol. 2006, 44, 265–274.
- Furuya, M. Complexity of tumor vasculature and molecular targeting therapies. Front. Biosci. 2011, 3, 549–561.
- Hida, K.; Maishi, N.; Torii, C.; Hida, Y. Tumor angiogenesis—characteristics of tumor endothelial cells. Int. J. Clin. Oncol. 2016, 21, 206–212.
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684.
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736.
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1α. Oncogene 2009, 28, 3949–3959.
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 1–16.
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284.
- Heddleston, J.M.; Li, Z.; Lathia, J.D.; Bao, S.; Hjelmeland, A.B.; Rich, J.N. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 2010, 102, 789–795.
- Ricciuti, B.; Foglietta, J.; Bianconi, V.; Sahebkar, A.; Pirro, M. Enzymes involved in tumor-driven angiogenesis: A valuable target for anticancer therapy. Semin. Cancer Boil. 2019, 56, 87–99.
- Jeong, H.; Kim, S.; Hong, B.-J.; Lee, C.-J.; Kim, Y.-E.; Bok, S.; Oh, J.-M.; Gwak, S.-H.; Yoo, M.Y.; Lee, M.S.; et al. Tumor-associated macrophages enhance tumor hypoxia and aerobic glycolysis. Cancer Res. 2019.
- Tanriover, G.; Aytac, G. Mutualistic Effects of the Myeloid-Derived Suppressor Cells and Cancer Stem Cells in the Tumor Microenvironment. Crit. Rev. Oncog. 2019, 24, 61–67.
- Guo, Y.; Feng, K.; Wang, Y.; Han, W. Targeting cancer stem cells by using chimeric antigen receptor-modified T cells: A potential and curable approach for cancer treatment. Protein Cell 2017, 9, 516–526.
- Miele, L.; Espinoza, I.; Pochampally, R.R.; Watabe, K.; Xing, F. Notch signaling: Targeting cancer stem cells and epithelial-to-mesenchymal transition. OncoTargets Ther. 2013, 6, 1249–1259.
- He, S.; Liu, Z.; Oh, D.-Y.; Thiele, C.J. MYCN and the epigenome. Front. Oncol. 2013, 3, 1.
- Guo, Z.; Jiang, J.-H.; Zhang, J.; Yang, H.-J.; Yang, F.-Q.; Qi, Y.-P.; Zhong, Y.-P.; Su, J.; Yang, R.-R.; Li, L.-Q.; et al. COX-2 Promotes Migration and Invasion by the Side Population of Cancer Stem Cell-Like Hepatocellular Carcinoma Cells. Medicine 2015, 94, e1806.
- D’Angelo, R.C.; Ouzounova, M.; Davis, A.; Choi, D.; Tchuenkam, S.M.; Kim, G.; Luther, T.; Quraishi, A.A.; Şenbabaoğlu, Y.; Conley, S.J.; et al. Notch reporter activity in breast cancer cell lines identifies a subset of cells with stem cell activity. Mol. Cancer Ther. 2015, 14, 779–787.
- Lee, S.H.; Do, S.I.; Lee, H.J.; Kang, H.J.; Koo, B.S.; Lim, Y.C. Notch1 signaling contributes to stemness in head and neck squamous cell carcinoma. Lab. Investig. 2016, 96, 508–516.
- Hawley, T.S.; Riz, I.; Yang, W.; Wakabayashi, Y.; DePalma, L.; Chang, Y.-T.; Peng, W.; Zhu, J.; Hawley, R. Identification of an ABCB1 (P-glycoprotein)-positive carfilzomib-resistant myeloma subpopulation by the pluripotent stem cell fluorescent dye CDy1. Am. J. Hematol. 2013, 88, 265–272.
- Fender, A.W.; Nutter, J.M.; Fitzgerald, T.L.; Bertrand, F.E.; Sigounas, G. Notch-1 Promotes Stemness and Epithelial to Mesenchymal Transition in Colorectal Cancer. J. Cell. Biochem. 2015, 116, 2517–2527.
- Benedito, R.; Roca, C.; Sörensen, I.; Adams, S.; Gossler, A.; Fruttiger, M.; Adams, R.H. The Notch Ligands Dll4 and Jagged1 Have Opposing Effects on Angiogenesis. Cell 2009, 137, 1124–1135.
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337.
- Owen, J.L.; Zadeh, M. Macrophages and chemokines as mediators of angiogenesis. Front. Physiol. 2013, 4, 159.
- Meier, K.; Lehr, C.-M.; Daum, N. Differentiation potential of human pancreatic stem cells for epithelial- and endothelial-like cell types. Ann. Anat. Anat. Anz. 2009, 191, 70–82.
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.W.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833.
- Grillet, F.; Bayet, E.; Villeronce, O.; Zappia, L.; Lagerqvist, E.L.; Lunke, S.; Charafe-Jauffret, E.; Pham, K.; Molck, C.; Rolland, N.; et al. Circulating tumour cells from patients with colorectal cancer have cancer stem cell hallmarks in ex vivo culture. Gut 2017, 66, 1802–1810.
- Burgess, D.J. Breast cancer: Circulating and dynamic EMT. Nat. Rev. Cancer 2013, 13, 148–149.
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449.
- Zhao, Y.; Bao, Q.; Renner, A.; Camaj, P.; Eichhorn, M.; Ischenko, I.; Angele, M.; Kleespies, A.; Jauch, K.W.; Bruns, C.J. Cancer stem cells and angiogenesis. Int. J. Dev. Boil. 2011, 55, 477–482.
- Bao, S. Stem Cell-like Glioma Cells Promote Tumor Angiogenesis through Vascular Endothelial Growth Factor. Cancer Res. 2006, 66, 7843–7848.
- Monzani, E.; Facchetti, F.; Galmozzi, E.; Corsini, E.; Benetti, A.; Cavazzin, C.; Gritti, A.; Piccinini, A.; Porro, D.; Santinami, M.; et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur. J. Cancer 2007, 43, 935–946.
- Maeda, S.; Shinchi, H.; Kurahara, H.; Mataki, Y.; Maemura, K.; Sato, M.; Natsugoe, S.; Aikou, T.; Takao, S. CD133 expression is correlated with lymph node metastasis and vascular endothelial growth factor-C expression in pancreatic cancer. Br. J. Cancer 2008, 98, 1389–1397.
- Gao, H.; Chakraborty, G.; Lee-Lim, A.P.; Mo, Q.; Decker, M.; Vonica, A.; Shen, R.; Brogi, E.; Brivanlou, A.H.; Giancotti, F.G. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 2012, 150, 764–779.
- Giancotti, F.G. Mechanisms Governing Metastatic Dormancy and Reactivation. Cell 2013, 155, 750–764.
- Kemper, K.; Grandela, C.; Medema, J.P. Molecular identification and targeting of colorectal cancer stem cells. Oncotarget 2010, 1, 387–395.
- Zhang, J.; Yuan, B.; Zhang, H.; Li, H. Human epithelial ovarian cancer cells expressing CD105, CD44 and CD106 surface markers exhibit increased invasive capacity and drug resistance. Oncol. Lett. 2019, 17, 5351–5360.
- Liu, J.; Chen, L.; Deng, H.; Xu, B.; Li, M.; Zheng, X.; Wu, C.; Jiang, J.-T. Epithelial-to-mesenchymal transition in human esophageal cancer associates with tumor progression and patient’s survival. Int. J. Clin. Exp. Pathol. 2014, 7, 6943–6949.
- Calon, A.; Lonardo, E.; Berenguer, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.F.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329.
- Chang, J.C. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine (Baltimore) 2016, 95, S20–S25.
- Hermann, P.C.; Sainz, B. Pancreatic cancer stem cells: A state or an entity? Semin. Cancer Boil. 2018, 53, 223–231.
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 Is a Marker of Constitutive and Reprogrammed Cancer Stem Cells Driving Colon Cancer Metastasis. Cell Stem Cell 2014, 14, 342–356.
- Yang, Z.; Ni, W.; Cui, C.; Qi, W.; Piao, L.; Xuan, Y. Identification of LETM1 as a marker of cancer stem-like cells and predictor of poor prognosis in esophageal squamous cell carcinoma. Hum. Pathol. 2018, 81, 148–156.
- Gupta, P.B.; Önder, T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659.
- Bure, I.V.; Nemtsova, M.V.; Zaletaev, D.V. Roles of E-cadherin and Noncoding RNAs in the Epithelial–mesenchymal Transition and Progression in Gastric Cancer. Int. J. Mol. Sci. 2019, 20, 2870.
- Zhou, Z.; Zhang, H.-S.; Liu, Y.; Zhang, Z.-G.; Du, G.-Y.; Li, H.; Yu, X.-Y.; Huang, Y. Loss of TET1 facilitates DLD1 colon cancer cell migration via H3K27me3-mediated down-regulation of E-cadherin. J. Cell. Physiol. 2017, 233, 1359–1369.
- Wakefield, L.M.; Hill, C.S. Beyond TGFβ: Roles of other TGFβ superfamily members in cancer. Nat. Rev. Cancer 2013, 13, 328–341.
- Parbin, S.; Kar, S.; Shilpi, A.; Sengupta, D.; Deb, M.; Rath, S.K.; Patra, S.K. Histone deacetylases: A saga of perturbed acetylation homeostasis in cancer. J. Histochem. Cytochem. 2014, 62, 11–33.
- Ganai, S.A. Histone deacetylase inhibitors modulating non-epigenetic players: The novel molecular targets for therapeutic intervention. Curr. Drug Targets 2018, 17, 1.
- Cao, Q.; Yu, J.; Han, S.; Kim, J.H.; Mani, R.-S.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Cao, X.; Kleer, C.G.; et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008, 27, 7274–7284.
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630.
- Calon, A.; Tauriello, D.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Boil. 2014, 25, 15–22.
- Park, S.-M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907.
- Guan, T.; Dominguez, C.X.; Amezquita, R.A.; Laidlaw, B.J.; Cheng, J.; Henao-Mejia, J.; Williams, A.; Flavell, R.A.; Lu, J.; Kaech, S.M. ZEB1, ZEB2, and the miR-200 family form a counterregulatory network to regulate CD8+ T cell fates. J. Exp. Med. 2018, 215, 1153–1168.
- Tellez, C.S.; Juri, D.E.; Do, K.; Bernauer, A.M.; Thomas, C.L.; Damiani, L.A.; Tessema, M.; Leng, S.; Belinsky, S.A. EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells. Cancer Res. 2011, 71, 3087–3097.
- Menéndez, J.; Alarcón, T. Metabostemness: A New Cancer Hallmark. Front. Oncol. 2014, 4, 262.
- Lee, K.-M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sánchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e7.
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794.
- Chiche, J.; Ilc, K.; Laferrière, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouyssegur, J. Hypoxia-Inducible Carbonic Anhydrase IX and XII Promote Tumor Cell Growth by Counteracting Acidosis through the Regulation of the Intracellular pH. Cancer Res. 2009, 69, 358–368.
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866.
- Nakada, D. Venetolax with Azacitidine Drains Fuel from AML Stem Cells. Cell Stem Cell 2019, 24, 7–8.
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Mutti, L.; Krstic-Demonacos, M.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 2016, 7, 34084–34099.
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4.
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37.
- Amsalem, Z.; Arif, T.; Shteinfer-Kuzmine, A.; Chalifa-Caspi, V.; Shoshan-Barmatz, V. The Mitochondrial Protein VDAC1 at the Crossroads of Cancer Cell Metabolism: The Epigenetic Link. Cancers 2020, 12, 1031.
- Skoda, J.; Borankova, K.; Jansson, P.; Huang, M.L.-H.; Veselska, R.; Richardson, D.R. Pharmacological targeting of mitochondria in cancer stem cells: An ancient organelle at the crossroad of novel anti-cancer therapies. Pharmacol. Res. 2019, 139, 298–313.
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312.
- Song, I.-S.; Jeong, Y.J.; Jeong, S.H.; Heo, H.J.; Kim, H.K.; Bae, K.B.; Park, Y.-H.; Kim, S.-U.; Kim, J.-M.; Kim, N.; et al. FOXM1-Induced PRX3 Regulates Stemness and Survival of Colon Cancer Cells via Maintenance of Mitochondrial Function. Gastroenterology 2015, 149, 1006–1016.e9.
- Isayev, O.; Rausch, V.; Bauer, N.; Liu, L.; Fan, P.; Zhang, Y.; Gladkich, J.; Nwaeburu, C.C.; Mattern, J.; Mollenhauer, M.; et al. Inhibition of glucose turnover by 3-bromopyruvate counteracts pancreatic cancer stem cell features and sensitizes cells to gemcitabine. Oncotarget 2014, 5, 5177–5189.
- Arif, T.; Paul, A.; Krelin, Y.; Shteinfer-Kuzmine, A.; Shoshan-Barmatz, V. Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States. Cancers 2018, 10, 499.
- Arif, T.; Krelin, Y.; Nakdimon, I.; Benharroch, D.; Paul, A.; Dadon-Klein, D.; Shoshan-Barmatz, V. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro Oncol. 2017, 19, 951–964.
- Dawson, M.A. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science 2017, 355, 1147–1152.
- Chatterjee, A.; Rodger, E.J.; Eccles, M. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin. Cancer Boil. 2018, 51, 149–159.
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu. Rev. Med. 2016, 67, 73–89.
- Borley, J.; Brown, R. Epigenetic mechanisms and therapeutic targets of chemotherapy resistance in epithelial ovarian cancer. Ann. Med. 2015, 47, 359–369.
- Arrowsmith, C.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400.
- Gates, L.A.; Shi, J.; Rohira, A.D.; Feng, Q.; Zhu, B.; Bedford, M.T.; Sagum, C.A.; Jung, S.Y.; Qin, J.; Tsai, M.-J.; et al. Acetylation on histone H3 lysine 9 mediates a switch from transcription initiation to elongation. J. Boil. Chem. 2017, 292, 14456–14472.
- Wolny, E.; Braszewska-Zalewska, A.; Kroczek, D.; Hasterok, R. Histone H3 and H4 acetylation patterns are more dynamic than those of DNA methylation in Brachypodium distachyon embryos during seed maturation and germination. Protoplasma 2017, 254, 2045–2052.
- Cruz, C.; Della Rosa, M.; Krueger, C.; Gao, Q.; Horkai, D.; King, M.; Field, L.; Houseley, J. Tri-methylation of histone H3 lysine 4 facilitates gene expression in ageing cells. eLife 2018, 7, 34081.
- Huang, H.; Weng, H.; Zhou, K.; Wu, T.; Zhao, B.S.; Sun, M.; Chen, Z.; Deng, X.; Xiao, G.; Auer, F.; et al. Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature 2019, 567, 414–419.
- Li, F.; Wu, R.; Cui, X.; Zha, L.; Yu, L.; Shi, H.; Xue, B. Histone Deacetylase 1 (HDAC1) Negatively Regulates Thermogenic Program in Brown Adipocytes via Coordinated Regulation of Histone H3 Lysine 27 (H3K27) Deacetylation and Methylation. J. Boil. Chem. 2016, 291, 4523–4536.
- Möller, M.; Schotanus, K.; Soyer, J.L.; Haueisen, J.; Happ, K.; Stralucke, M.; Happel, P.; Smith, K.M.; Connolly, L.R.; Freitag, M.; et al. Destabilization of chromosome structure by histone H3 lysine 27 methylation. PLoS Genet. 2019, 15, e1008093.
- Bird, A. CpG-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213.
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45.
- Cosgrove, M.S.; Wolberger, C. How does the histone code work? Biochem. Cell. Biol. 2005, 83, 468–476.
- Cloney, R. Gene regulation: Optical control of epigenetics. Nat. Rev. Genet. 2016, 17, 254.
- Harvey, Z.; Chen, Y.; Jarosz, D.F. Protein-Based Inheritance: Epigenetics beyond the Chromosome. Mol. Cell 2017, 69, 195–202.
- Kim, M.Y.; Lee, J.E.; Kim, L.K.; Kim, T. Epigenetic memory in gene regulation and immune response. BMB Rep. 2019, 52, 127–132.
- Goldman, M. CpG Islands. In Encyclopedia of Genetics; Elsevier BV: Amsterdam, The Netherlands, 2001; p. 477.
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022.
- Ohtani-Fujita, N.; Fujita, T.; Aoike, A.; Osifchin, N.E.; Robbins, P.D.; Sakai, T. CpG methylation inactivates the promoter activity of the human retinoblastoma tumor-suppressor gene. Oncogene 1993, 8, 8.
- Di Croce, L.; Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Boil. 2013, 20, 1147–1155.
- Morey, L.; Helin, K. Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci. 2010, 35, 323–332.
- Corso-Díaz, X.; Jaeger, C.; Chaitankar, V.; Swaroop, A. Epigenetic control of gene regulation during development and disease: A view from the retina. Prog. Retin. Eye Res. 2018, 65, 1–27.
- Collinson, A.; Collier, A.; Morgan, N.P.; Sienerth, A.; Chandra, T.; Andrews, S.; Rugg-Gunn, P.J. Deletion of the Polycomb-Group Protein EZH2 Leads to Compromised Self-Renewal and Differentiation Defects in Human Embryonic Stem Cells. Cell Rep. 2016, 17, 2700–2714.
- Wainwright, E.N.; Scaffidi, P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer 2017, 3, 372–386.
- Donaldson, M.C.; Sungalee, S.; Zufferey, M.; Tavernari, D.; Katanayeva, N.; Battistello, E.; Mina, M.; Douglass, K.M.; Rey, T.; Raynaud, F.; et al. EZH2 oncogenic mutations drive epigenetic, transcriptional, and structural changes within chromatin domains. Nat. Genet. 2019, 51, 517–528.
- Woods, B.A.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Immunol. Rev. 2014, 263, 22–35.
- Yi, J.M.; Tsai, H.-C.; Glöckner, S.C.; Lin, S.; Ohm, J.E.; Easwaran, H.; James, C.D.; Costello, J.F.; Riggins, G.; Eberhart, C.G.; et al. Abnormal DNA methylation of CD133 in colorectal and glioblastoma tumors. Cancer Res. 2008, 68, 8094–8103.
- Tabu, K.; Sasai, K.; Kimura, T.; Wang, L.; Aoyanagi, E.; Kohsaka, S.; Tanino, M.; Nishihara, H.; Tanaka, K. Promoter hypomethylation regulates CD133 expression in human gliomas. Cell Res. 2008, 18, 1037–1046.
- Gorodetska, I.V.; Lukiyanchuk, V.; Peitzsch, C.; Kozeretska, I.; Dubrovska, A. BRCA1 and EZH2 cooperate in regulation of prostate cancer stem cell phenotype. Int. J. Cancer 2019, 145, 2974–2985.
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715.
- Cai, C.; Zhu, X. The Wnt/beta-catenin pathway regulates self-renewal of cancer stem-like cells in human gastric cancer. Mol. Med. Rep. 2012, 5, 1191–1196.
- Matsui, W.H. Cancer stem cell signaling pathways. Medicine 2016, 95, S8–S19.
- Zhu, C.; Pan, Y.; Ma, S.; Cao, K.; Zhou, S.; Zhao, A.; Li, M.; Qian, F. Therapeutic approaches targeting cancer stem cells. J. Cancer Res. Ther. 2018, 14, 1469.
- Lai, E. Notch signaling: Control of cell communication and cell fate. Development 2004, 131, 965–973.
- Dessaud, E.; McMahon, A.P.; Briscoe, J. Pattern formation in the vertebrate neural tube: A sonic hedgehog morphogen-regulated transcriptional network. Development 2008, 135, 2489–2503.
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464.
- Zhang, J.; Shao, X.; Sun, H.; Liu, K.; Ding, Z.; Chen, J.; Fang, L.; Su, W.; Hong, Y.; Li, H.; et al. NUMB negatively regulates the epithelial-mesenchymal transition of triple-negative breast cancer by antagonizing Notch signaling. Oncotarget 2016, 7, 61036–61053.
- Polakis, P. The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 2007, 17, 45–51.
- Gujral, T.S.; Chan, M.; Peshkin, L.; Sorger, P.K.; Kirschner, M.W.; MacBeath, G. A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell 2014, 159, 844–856.
- Yang, L.; Xie, G.; Fan, Q.; Xie, J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene 2009, 29, 469–481.
- Clement, V.; Sanchez, P.; De Tribolet, N.; Radovanovic, I.; i Altaba, A.R. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172.
- Steinway, S.N.; Zañudo, J.G.; Ding, W.; Rountree, C.B.; Feith, D.J.; Loughran, T.P.; Albert, R. Network Modeling of TGF Signaling in Hepatocellular Carcinoma Epithelial-to-Mesenchymal Transition Reveals Joint Sonic Hedgehog and Wnt Pathway Activation. Cancer Res. 2014, 74, 5963–5977.
- Fan, H.-X.; Wang, S.; Zhao, H.; Liu, N.; Chen, D.; Sun, M.; Zheng, J. Sonic hedgehog signaling may promote invasion and metastasis of oral squamous cell carcinoma by activating MMP-9 and E-cadherin expression. Med. Oncol. 2014, 31, 1–8.
- Suzuki, H.; Watkins, D.N.; Jair, K.-W.; Schuebel, K.E.; Markowitz, S.D.; Chen, W.D.; Pretlow, T.P.; Yang, B.; Akiyama, Y.; Van Engeland, M.; et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat. Genet. 2004, 36, 417–422.
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60.
- Hussain, M.; Rao, M.; Humphries, A.E.; Hong, J.A.; Liu, F.; Yang, M.; Caragacianu, D.; Schrump, D.S. Tobacco Smoke Induces Polycomb-Mediated Repression of Dickkopf-1 in Lung Cancer Cells. Cancer Res. 2009, 69, 3570–3578.
- Ghoshal, P.; Nganga, A.J.; Moran-Giuati, J.; Szafranek, A.; Johnson, T.R.; Bigelow, A.J.; Houde, C.M.; Avet-Loiseau, H.; Smiraglia, D.J.; Ersing, N.; et al. Loss of the SMRT/NCoR2 Corepressor Correlates with JAG2 Overexpression in Multiple Myeloma. Cancer Res. 2009, 69, 4380–4387.
- Wu, Y.; Gong, L.; Xu, J.; Mou, Y.; Xu, X.; Qian, Z. The clinicopathological significance of HES1 promoter hypomethylation in patients with colorectal cancer. OncoTargets Ther. 2017, 10, 5827–5834.
- Benoit, Y.D.; Laursen, K.B.; Witherspoon, M.S.; Lipkin, S.M.; Gudas, L.J. Inhibition of PRC2 histone methyltransferase activity increases TRAIL-mediated apoptosis sensitivity in human colon cancer cells. J. Cell. Physiol. 2013, 228, 764–772.
- Han, X.; Ranganathan, P.; Tzimas, C.; Weaver, K.L.; Jin, K.; Astudillo, L.; Zhou, W.; Zhu, X.; Li, B.; Robbins, D.J.; et al. Notch Represses Transcription by PRC2 Recruitment to the Ternary Complex. Mol. Cancer Res. 2017, 15, 1173–1183.
- Jagani, Z.; Mora-Blanco, E.L.; Sansam, C.G.; McKenna, E.S.; Wilson, B.; Chen, D.; Klekota, J.; Tamayo, P.; Nguyen, P.T.L.; Tolstorukov, M.; et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat. Med. 2010, 16, 1429–1433.
- McKenna, E.S.; Tamayo, P.; Cho, Y.-J.; Tillman, E.J.; Mora-Blanco, E.L.; Sansam, C.G.; Koellhoffer, E.C.; Pomeroy, S.L.; Roberts, C.W.M. Epigenetic inactivation of the tumor suppressor BIN1 drives proliferation of SNF5-deficient tumors. Cell Cycle 2012, 11, 1956–1965.
- Coni, S.; Mancuso, A.B.; Di Magno, L.; Sdruscia, G.; Manni, S.; Serrao, S.M.; Rotili, D.; Spiombi, E.; Bufalieri, F.; Petroni, M.; et al. Selective targeting of HDAC1/2 elicits anticancer effects through Gli1 acetylation in preclinical models of SHH Medulloblastoma. Sci. Rep. 2017, 7, 44079.
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell. Biol. 2010, 12, 132–142.
- Niewiadomski, P.; Niedziółka, S.M.; Markiewicz, Ł.; Uśpieński, T.; Baran, B.; Chojnowska, K. Gli Proteins: Regulation in Development and Cancer. Cells 2019, 8, 147.
- Michalak, E.M.; Visvader, J.E. Dysregulation of histone methyltransferases in breast cancer – Opportunities for new targeted therapies? Molecular Oncol. 2016, 10, 1497–1515.
- Rao, R.C.; Dou, Y. Hijacked in cancer: The KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer 2015, 15, 334–346.
- Cozzio, A.; Passegué, E.; Ayton, P.M.; Karsunky, H.; Cleary, M.L.; Weissman, I.L. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003, 17, 3029–3035.
- Ono, R.; Masuya, M.; Nakajima, H.; Enomoto, Y.; Miyata, E.; Nakamura, A.; Ishii, S.; Suzuki, K.; Shibata-Minoshima, F.; Katayama, N.; et al. Plzf drives MLL-fusion–mediated leukemogenesis specifically in long-term hematopoietic stem cells. Blood 2013, 122, 1271–1283.
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL–AF9. Nature 2006, 442, 818–822.
- Somervaille, T.C.; Matheny, C.J.; Spencer, G.J.; Iwasaki, M.; Rinn, J.; Witten, D.M.; Chang, H.Y.; Shurtleff, S.A.; Downing, J.R.; Cleary, M.L. Hierarchical Maintenance of MLL Myeloid Leukemia Stem Cells Employs a Transcriptional Program Shared with Embryonic Rather Than Adult Stem Cells. Cell Stem Cell 2009, 4, 129–140.
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 Activity by a Gain-of-Function H3 Mutation Found in Pediatric Glioblastoma. Science 2013, 340, 857–861.
- Benetatos, L.; Vartholomatos, G. On the potential role of DNMT1 in acute myeloid leukemia and myelodysplastic syndromes: Not another mutated epigenetic driver. Ann. Hematol. 2016, 95, 1571–1582.
- Brunetti, L.; Gundry, M.C.; A Goodell, M. DNMT3A in Leukemia. Cold Spring Harb. Perspect. Med. 2016, 7, a030320.
- Sun, Y.; Chen, B.-R.; Deshpande, A. Epigenetic Regulators in the Development, Maintenance, and Therapeutic Targeting of Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 41.
- Humphries, H.N.; Wickremesekera, S.K.; Marsh, R.W.; Brasch, H.D.; Mehrotra, S.; Tan, S.T.; Itinteang, T. Characterization of Cancer Stem Cells in Colon Adenocarcinoma Metastasis to the Liver. Front. Surg. 2018, 4, 76.
- Munro, M.; Wickremesekera, S.K.; Peng, L.; Tan, S.T.; Itinteang, T. Cancer stem cells in colorectal cancer: A review. J. Clin. Pathol. 2017, 71, 110–116.
- Todaro, M.; Francipane, M.G.; Medema, J.P.; Stassi, G. Colon Cancer Stem Cells: Promise of Targeted Therapy. Gastroenterology 2010, 138, 2151–2162.
- Hammoud, S.S.; Cairns, B.R.; Jones, D.A. Epigenetic regulation of colon cancer and intestinal stem cells. Curr. Opin. Cell Boil. 2013, 25, 177–183.
- Campbell, P.M.; Szyf, M. Human DNA methyltransferase gene DNMT1 is regulated by the APC pathway. Carcinog. 2003, 24, 17–24.
- Liang, T.-J.; Wang, H.-X.; Zheng, Y.-Y.; Cao, Y.-Q.; Wu, X.; Zhou, X.; Dong, S.-X. APC hypermethylation for early diagnosis of colorectal cancer: A meta-analysis and literature review. Oncotarget 2017, 8, 46468–46479.
- Paschall, A.V.; Liu, K. Epigenetic and Immune Regulation of Colorectal Cancer Stem Cells. Curr. Color. Cancer Rep. 2015, 11, 414–421.
- Pathania, R.; Ramachandran, S.; Elangovan, S.; Padia, R.; Yang, P.; Cinghu, S.; Veeranan-Karmegam, R.; Arjunan, P.; Gnana-Prakasam, J.P.; Sadanand, F.; et al. DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat. Commun. 2015, 6, 1–11.