Introduction
Atmospheric processes are a blend of meteorological phenomena, air chemistry and interactions with the hydro-, pedo- and biosphere. Natural chemical processes are further complicated by the addition of anthropogenic gas and particle sources emitted into the atmosphere. Contemporary reductionist philosophy seeks separation of these components for a simplifying strategy to characterize parts of widely different elements of atmospheric phenomena in time and space. Research strategies have applied separate, fundamental results of physics, chemistry and biology to construct conceptual models leading to current understanding of the atmospheric chemical dynamics. An adjunct to these applications includes laboratory simulation of different processes believed to be relevant to the atmosphere [1]. Contemporary simulation technology has recognized the spatial and temporal scale of atmospheric processes from the microscale, including chemical kinetics and cloud micro-physics, to planetary scale fluid dynamics of rotating stratified fluids.
Simulation of processes indirectly relevant to atmospheric chemistry began with laboratory experiments of J. Tyndall in 1868 [2] followed with C.T.R. Wilson’s 1897 cloud chamber studies [3]. Tyndall experimented with particle production in irradiated gases in a tubular reactor. The gases exposed to light revealed the potential for photochemical processes, perhaps the first hint of secondary aerosol production possible from irradiated trace species in air. Wilson’s experiments elucidated the nature of homogeneous nucleation in supersaturated vapors and found the mediating effects lowering supersaturation that were required for vapor condensation by ions. These experiments partially formed the basis for several atmospheric studies of ions, nuclei formation and cloud formation.
Physicochemical phenomena in the atmosphere depend on the interactions of processes involving the atmosphere’s energy balance and distribution, fluid dynamics, cloud physics and chemistry, and underlying (heterogeneous) surface exchange. Expanding knowledge of photochemical processes in the atmosphere called attention to the potential for experimentally separating chemical processes from these other processes. The genesis of simulating tropospheric photochemistry began with suspicions of the presence of oxidants (ozone) in the Los Angeles atmosphere [4]. In the 1950s, Haagen-Smit’s [5] famous 2-liter flask experiments surfaced confirming the presence of ozone (O3) in smog, and empirically exploring the role of nitrogen oxides (NOx = NO + NO2) and hydrocarbons (HC) in oxidant chemistry. These studies aroused the regulatory concern of both the local pollution control authorities and industrial or motor vehicles representatives leading early on to the creation of a cooperative research entity in 1954, the Air Pollution Foundation. Renzetti’s review [6] describes a succession of early laboratory experiments following up on Haagen-Smit’s experiments. Subsequent studies of the smog photochemistry expanded in chambers up to 50 liters volume. In 1961 a major, sustained US commitment to gas-phase smog chamber studies was implemented at the University of California Riverside (UCR) Statewide Air Pollution Research Center (SAPRC, later renamed APRC). This initiative fostered the design and development of major smog chamber facilities.
A complementary concern from urban observations of smog was the presence of suspended particles leading to severe visibility impairment and the potential for exacerbation of ischemic or specific respiratory disease [7]. Particle concentrations following the diurnal changes in photochemical smog pointed to a chemical link distinct from smoke or dust emissions. This conjecture was verified early on in laboratory experiments of Renzetti and colleagues [8,9,10], who irradiated auto exhaust in a reactor with and without sulfur dioxide (SO2). They found that SO2 was readily oxidized by photochemical oxidants with water to form sulfuric acid along with organic material. Earlier, Mader et al. [11] found organic particles in filtered smoggy Los Angeles air, along with sulfate. The presence of sulfur oxides in polluted air and nucleation forming H2SO4 or sulfate salts in the 1950s followed a long history of knowledge dating back centuries [12].
Atmospheric measurements showing photochemical phenomena motivated investigators to begin studying aerosol production in laboratory photochemical systems after the 1960s [13-17]. Photochemical aerosol studies initially focused on sulfate [18-21]. Later, simulation experiments on organic carbon particles began to appear, for example, [22-25]. Most of these experiments were conducted in batch photochemical reactors or environmental (smog) chambers that were operated at various institutions.
Following the photochemical research of the 1950s [26], the mechanisms responsible for oxidant formation was hypothesized to involve the O3-NOx inorganic cycle often attributed to Chapman [27] combined with free radical interactions of reactive nitrogen (NOy) and non-methane hydrocarbons (part of volatile organic compounds--VOCs). The free radical chemistry, especially for hydroxyl radical (OH), was determined to be critical in chain reactions leading to maintaining oxidant concentrations beyond the Chapman cycle.
Conceptually, the rapidly evolving post-1960s photochemical kinetic studies fostered the interest in using smog chambers as a means of study of atmospheric photochemistry. Simulating microscale processes relevant to the atmosphere included the complex chemistry of trace gases and aerosol particles in the air [28, 29]. In the 1960s, the studies of gas-phase and aerosol chemistry paralleled one another. Gas-phase photochemical processes involving NOx and VOC relevant to urban conditions took precedence through the 1980s with combined bench scale experiments and smog chamber research actively pursued at the at SAPRC, for example, and later at the University of North Carolina, Carnegie Mellon University and the US Environmental Protection Agency (EPA). Experiments for aerosol photochemistry took place using indoor or outdoor chambers at SAPRC (later the UCR Center for Environmental Research and Technology, CE-CERT), the California Institute of Technology and Carnegie Mellon University. These paralleled similar research at the University of North Carolina, and in Europe and Asia [30-35].
Smog chamber experiments have complemented laboratory chemical kinetics research and enabled the verification of various smog chemistry mechanisms for air quality models, as well as for the exploration of source emission controls on photochemistry in different environmental conditions [28, 36]. Aerosol chamber studies have advanced the knowledge of secondary particle production from gaseous species in different environments [37].
The goal of this narrative is to first summarize the technical capabilities of environmental simulation chambers required for experimentation, and other studies that have qualified chamber results relevant to the atmosphere. A survey is given of chamber facilities operated throughout the early 21st century. To illustrate the importance of chamber research, a summary is provided for some of the many contemporary contributions to knowledge of smog chemistry resulting from these experiments. Atmospheric relevance of smog chambers for kinetics studies and as simulators end the narrative. The details of research leading to various gas-phase photochemical-aerosol mechanisms are left to the wisdom derived from many reviews in the literature exemplified in Table 1.
Table 1. Some reviews of studies relevant to atmospheric chemistry (2000–2019).
|
Review |
Year |
Content |
|
Finlayson-Pitts and Pitts [28] |
2000 |
Textbook-comprehensive review of gas and aerosol chemistry relevant to the atmosphere through the 1990s.
|
|
Dodge [36] |
2000 |
Atmospheric chemistry of O3 with analysis of smog chambers and their results.
|
|
Atkinson[38] |
2000 |
Photochemical mechanism review with relevance to atmospheric chemistry.
|
|
George et al. [39] |
2005 |
Comprehensive review of heterogeneous atmospheric chemistry
|
|
Seinfeld and Pandis (3rd ed.) [29] |
2006 |
Textbook-comprehensive gas and aerosol chemistry with summary of meteorological and climate relevance through early 2000s.
|
|
Kroll and Seinfeld [40] |
2008 |
Review of photochemical aerosol formation with reference to smog chamber studies through mid- 2000s.
|
|
Hallquist et al. [37] |
2009 |
Review of secondary organic aerosol (SOA) formation including photochemical processes.
|
|
Carlton et al., [41] |
2009 |
Secondary organic aerosols (SOA) from isoprene oxidation.
|
|
Lim et al. [42] |
2010 |
Aqueous chemistry and formation of SOA.
|
|
Stockwell et al. [43] |
2012 |
Chemical mechanisms and models for O3.
|
|
Glasius and Goldstein [44] |
2016 |
Review of contemporary atmospheric organic chemistry status and future emphasizing SOA.
|
|
Bianchi et al. [45] |
2019 |
Review of autoxidation and highly oxygenated organic molecules (HOM) in atmospheric chemistry.
|
Requirements for Chamber Technology
Simulation of atmospheric chemical processes involves separation of an array of interactions that result in ambient concentrations of gaseous and particulate species. These interactions involve phenomena that range of time scales from a few seconds to several days, and spatial scales from less than a cm to km’s. They include rapid homogeneous or heterogeneous reaction pathways superimposed on a “background” of ever-present trace species concentrations, the intermittent presence of aerosols and hydrometeor clouds, contaminant emissions, dilution via mixing of air with different properties. Simulation schemes obviously apply to conditions favoring extremely small time and space constraints. They are designed to isolate “pure” chemical phenomena from the many other phenomena taking place in the atmosphere and the atmospheric boundaries. This approach asserts that such a separation leads to descriptions from building blocks relevant to the atmosphere; in so doing, they advance our understanding of the atmosphere, and enable a predictive capability from integrated process modeling (see also Section 4).
The study of atmospheric photochemistry derives from observations and early experiments of Haagen-Smit and colleagues, and scientists affiliated with the Los Angeles Air Pollution Control District. These studies focused on the NOx-VOC system, air pollutant agents from a variety of sources. Early on, a decision was made to separate the gas-phase processes from aerosol processes even though they were obviously observationally coupled in the atmosphere. The separation of gas and particle processes in the 1950s evidently followed from a conclusion that visibility and haze were connected to smoke sources distinct from photochemical reactions. By the 1970s the coupling between haze and gas-phase photochemistry was widely recognized as an issue in characterizing smog.
Observations of the atmospheric NOx-VOC photochemical cycle showed that simulation would require several hours to trace the chemical reactions to some end point past a maximum oxidant concentration. This precluded most laboratory designs that focused on kinetics on a scale of minutes or less even though flow reactors with short gas residence times 100–250 seconds have been used to obtain chemical information relevant to the atmosphere [46-49].
The value of chamber simulations lies with the ability to measure under controlled conditions the complex chemical kinetics taking place in as much detail as instrumentation will permit. Since the 1950s, gas-phase measurement technologies have improved dramatically for extracted samples as well as continuous in situ observations. Advances in measurement methods, including spectroscopy and chromatography, have supplanted early use of wet chemical methods and Haagen-Smit’s [5] rubber cracking rate for ozone (O3) concentration changes. Continuous aerosol particle measurement capability has followed gas-phase technology closely in recent decades, complementing extractive methods of substrate sampling and batch chemical analysis.
Ideal Design Considerations
A high-fidelity simulation of atmospheric photochemistry requires that several conditions be considered. These include:
- Initial and endpoint concentrations of reactive species should approach those observed in the atmosphere.
- Initial input of gas mixtures (or aerosols) should be well-defined so that spurious reactions caused by reactant contamination are controlled entering the vessel.
- The light source for irradiation should simulate sunlight, or at a minimum, the light wavelengths known to be key for excitation of airborne species.
- The duration of an experiment should approach at least most of a day or a span of several hours.
- Air in the chambers should be well mixed to minimize concentration gradients in the chamber.
- Documentation and control of environmental variables like pressure, humidity and temperature should be assured or variability at least documented.
- Loss of reactants, degassing or byproducts of surface reactions at the vessel walls should be accounted for as an influence on the measured reactant-product profiles.
- Instrumentation for characterizing reactions should be as complete as practical from speciated VOCs and NOy (reactive nitrogen species) to oxidants, and particle physicochemical characteristics or other stable products and inference of free radical or other intermediate species.
The designers of simulators have responded to these requirements with approaches adopting batch reactors (chambers) or flow reactors with nominally inert surfaces which are transparent to light below ~<450 nm wavelength. Chambers have been located either indoors as part laboratory capabilities or outdoors. They have been constructed of pyrex (borosilicate glass), quartz, stainless steel or Teflon films that are transparent to light to sunlight, particularly in the range of 200–450 nm wavelength, or artificial lighting from sources that simulate sunlight. Finlayson-Pitts and Pitts [28] summarize the pros and cons of these materials including their transparency to radiation and wall effects discussed below.
Simulation chambers have been deployed outdoors or indoors using light sources that include sunlight, black light from filtered mercury lamps, sun lamps, and xenon or argon arc lamps. Light intensities varying with configuration require measurement inside the chambers to document the wavelength coverage of the source, and require periodic verification of intensity compared with sunlight. Some of the indoor chambers include temperature and relative humidity (RH) control, and vacuum capability to expand the range of experiments accessible in the chamber. Outdoor chambers generally exclude temperature and RH control. Some chambers are capable of injecting water vapor as a means of RH variation during an experiment. Chambers have been operated as static vessels or dynamic systems with gas flow to simulate fresh reactant injection during the aging of the irradiated mixture.
The reactant and product concentrations need to be constrained in experiments such that the reactant gases are in the ppb-ppm range and O3 maxima are less than ~400 ppb and NOy should be similarly constrained. Free radical concentrations or stable intermediates in chambers will be far lower, challenging investigators to measure them either directly or inferring them through tracers. If particle formation is investigated, concentrations of 1–100 µg/m3 should be present as products. Desired measurements include continuous observation of total number concentration of particles and particle size distributions.
Concentrations of gases injected into chambers are historically higher than current urban atmospheric conditions, but are similar to observations in Los Angeles in the 1970s as an upper limit. The input gases for simulation chambers are blends containing pure NOx or mixtures of NOx and VOC with super-purified air to define the starting point of a sequence of reactions. Injection of gases can include outdoor air, source emissions such as internal combustion engines, or purified gas streams to investigate reactions associated with single VOC species or mixes that are similar to those from sources. Chambers are designed with the means of purifying inlet gases, including air, to remove NOx and VOC contamination to less than ppb levels.
Concentrations of NOx and VOC reactants are varied to achieve different VOC/NOx ratios in excess of 2. This ratio is well known to be an important parameter for establishing conditions near ambient, and limits of conditions of VOC or NOx sensitivity [28].
Studies of secondary organic aerosol (SOA) formation involve injection of specific VOC or mixtures of VOCs, beyond those usually adopted for oxidant formation. Also, the capability for adding “seed” aerosols for catalyzing new particle formation, or atmospherically representative particles to investigate the heterogeneous growth of particles. Other injection or generation capabilities include adjustment for humidity and “background or baseline” aerosols. During or after injection of the gas mixture needs to be well-mixed by fans or other methods to insure no gradients exist in the reactor volume.
Chambers are operated over time periods of a few hours to more than ten hours in various runs, simulating the early morning build-up of smog chemistry through midday and later. The later periods beyond the O3 maximum are problematic in the potential ambiguities of various reaction pathways occurring with declining sunlight and hourly “aging” of the gas or aerosol mixture. Multiday experiments with dilution have been attempted in outdoor chambers [50], but such experiments are subject to cumulative increasing ambiguities from wall effects and potential air stratification without mixing assistance.
Because of their importance for ensuring the fidelity of homogeneous chemistry believed to represent the atmosphere, two essential features need to be addressed: light sources and wall surface effects. Leighton and later Finlayson-Pitts and Pitts [26, 28] discussed the solar radiation spectrum in detail and the actinometric range of interest for tropospheric O3 chemistry. Finlayson-Pitts and Pitts, and others [28, 51, 52] summarized the various light sources used in smog chambers, including filtered UV lamps, sunlight, black light and xenon arc lamps. Light sources have different irradiation intensity distributions that constrain their simulation of the solar spectrum. For example, black light lamps are widely used because they are inexpensive and have a good UV cutoff at ~300nm. However, they do not represent the solar light intensity well in the visible regime, which is potentially important for photolysis [53]. Darnall et al. [54] noted the differences expected from constant and diurnal varying light. Light sources have been studied extensively to obtain chamber-specific data with light conditions to constrain the gas-phase reaction progress to take account of these variables.
Wall effects are of major concern for in tracing particle forming reactions in chambers. These effects not only involve the biases established from gas-phase NOx-VOC-oxidant reactions, but also interactions involving volatile and non-volatile products in the formation, coagulation or evaporation of aerosol particles, as well as physical removal of particles by deposition. For gas-phase systems, wall effects have been studied in detail to determine ways for correcting the observations for such effects. Dodge et al. [55] and Bufalini et al. [56] discussed the implications of contamination in chambers as a concern. Lonneman et al. [57] and Kelly et al. [58] investigated VOC contamination from Teflon films. Grosjean [59] noted wall losses as an uncertainty in chamber operations, and Yarwood et al. [60] examined NOx wall losses as part of more recent chamber experiments. Trump et al. [61] constructed a model for chamber losses. Empirically, the uncertainties associated with wall interactions can be addressed by baking out chambers [62], or treatment with dilute concentrations of O3.
Particularly concerning for atmospheric simulation is the potential for free radical interactions from chamber walls, especially the HONO disassociation to give a spurious OH supply. Carter et al. [63] hypothesized a release of free radicals from chamber walls with temperature and irradiation. Carter et al. [64,65] investigated this question further. Their studies were complemented with work of Besemer and Nieboer [66] and Sasamaki and Akimoto [67]. Rohrer et al. [68] noted the release of HONO from chamber walls as a continuing issue from the use of chambers. Iraci et al. [69] proposed a series of experiments that could add more information about gas-phase wall interactions.
As a practical measure, the influence of wall effects of deposition and adsorption–desorption of NO, NO2 and O3, or release of free radicals (e.g., OH from HONO reactions is said to be reduced by minimizing the surface to volume ratio of the chamber.). Empirical adjustments can be made to account for wall effects in gas-phase studies [61, 66]. Alternatively, investigators have designs employing low surface to volume ratios to minimize the influence of walls on the photochemistry.
Experiments of photochemical aerosol formation add complexity of particle interactions with chamber walls along with gas interactions. Aerosols formed in chambers can be lost by diffusion to the wall, or sedimentation of larger particles. Loss of charged particles to the walls from electrostatic attraction also has been documented [70,71]. Vapor and aerosol loss in secondary aerosol formation was discussed early on [72]. More recent studies of organic vapor-particle loss on Teflon surfaces have reported the potential for low bias in particle yields [73-78]. For low-volatility or semi-volatile organics, wall loss is complicated by mass transfer and reactions on seed or SOA surfaces, which can be accounted for empirically [79]. With the difficulty in comparing chambers and their results for wall-loss of organics, Brune [80] has proposed an empirical “index” for interrelating chamber conditions. Pierce et al. [81] proposed a theoretically-based model for loss from wall effects, coagulation and condensation-evaporation in chambers. Sunol et al. [82] presented a comprehensive model for characterizing secondary organic aerosol (SOA) formation in chambers, including accounting for wall effects.
Some Chamber Facilities
The design of smog chambers has taken into account the ideal requirements above in various ways to create practical containments for experimentation. The methodology adopted for design and conduct of experiments are discussed [17]. Since the 1950s, there are several smog chambers that have been constructed for both homogeneous gas-phase reactions and photochemical aerosol studies. These facilities have served a range of experiments for major efforts to develop well-documented data bases for mechanism studies and evaluation. The chambers available before 2000 for gas-phase studies are listed in Dodge [36]. The listing includes examples of both indoor and outdoor facilities with varying volumes and media, NOx levels and VOC selected for simulating polluted air. Dodge noted more than 1000 individual runs performed at her listed facilities have been available for developing or evaluating gas-phase oxidant forming mechanisms.
After the 1970s, smog chambers also were used to follow up earlier investigations of photochemical aerosol formation that were assumed to be relevant to the atmosphere. The post-1980s focused much more on secondary organic aerosols (SOAs) rather than the inorganic species, sulfate and nitrate. Table 2 gives a list of chambers for this application, which are located all over the world. These obviously could also be used for gas-phase kinetics studies as well. Table 2 includes the institution location, the volume of the container, light source, indoor-outdoor configuration, material of construction, operating temperature and references to their descriptions. They range in volume from less than 1 m3 to nearly 300 m3. Some of the facilities have dual (side-by-side) chambers that permit parallel experiments with differing input conditions.
Aerosol experiments have included not only NOx- oxidant-related VOC mixtures to produce oxidant, but have added a variety of aerosol-forming VOC (single and mixtures [8,83,84]) and SOx as SO2 - sulfuric acid to seek knowledge of the aerosol-forming kinetics expected in the atmosphere.
An example dual chamber facility constructed at the University of California Riverside is shown in Figure 1 [85].
Table 2. Some Smog Chambers Used for Photochemical Gas-Aerosol Studies (updated table from Hallquist et al. [37]).
|
Location |
Type |
Volume (m3) |
Material |
Temperature (oK) |
Referencea |
|
California Inst. of Technology |
Indoor photoreactor black light |
28 (dual) |
Teflon (FEP) |
290–303 |
Cocker [86];Ng [87] |
|
California Inst. of Technology |
Outdoor photoreactor |
65 |
FEP |
ambient |
Leone et al. [88] |
|
Carnegie Mellon Univ. |
Indoor photoreactor black light |
10 |
Teflon (PTFE/FEP) |
288-313 |
Stanier [89]; Robinson et al [90] |
|
Forschungazentrum Julich, Germany (SAPHIR) |
Outdoor photoreactor |
270 |
FEP |
ambient |
Rohrer et al [68] |
|
Forschungazentrum Julich, Germany |
Dark chamber |
250 |
PTFE/FEP |
ambient |
Mentel et al. [91]; Saathoff et al. [ 92] |
|
Forschungazentrum Karlsruhe, Germany (AIDA) |
Dark chamber (clouds) |
4-84 |
Metal |
183-323 |
Saathoff et al [93]; Jonsson et al. [94] |
|
Fundacion Centro de Estudios Ambientales del Mediterranean Spain (EUPHORE) |
Outdoor photoreactor |
200 |
FEP |
ambient |
Rodenas et al. [95]; Klotz et al. [96] |
|
Leibniz Institute for Tropospheric Research Germany |
Indoor photoreactor black light |
19 |
FEP |
289–308 |
Iinuma et al. [97] |
|
Paul Scherrer Insitute Switzerland |
Indoor photoreactor xenon arc |
27 |
FEP |
290–298 |
PSI [30]; Paulsen et al. [98] |
|
University College Cork Ireland |
Indoor photoreactor black light |
6.5 |
FEP |
293–305 |
Temime et al. [99]; Healy et al. [100] |
|
University of Manchester UK |
Indoor photoreactor Halogen/xenon arc |
18 |
FEP |
288–313 |
Centre for Atmos. Sci. [101] |
|
University of California Riverside (CECERT) US |
Indoor photoreacto xenon arc/black light |
90 (dual) |
FEP |
278–323 |
Carter et al. [85]; Song et al. [102] |
|
University of California Riverside (APRC) US |
Photoreactors (several indoor and outdoor ) xenon arc/black light |
6-8 |
PTFE/FEP |
ambient |
Dodge [36]; Tobias and Ziemann [103] |
|
University of North Carolina US |
Outdoor photoreactor |
120; 137 (dual); 150 (dual) |
FEP |
ambient |
Jeffries [104-106] ; Lee et al. [107] |
|
US EPA |
Indoor photoreactor Black light |
14.5 |
FEP/TFE |
293-298 |
Edney [108] |
|
CSIRO |
Outdoor photoreactor |
20 (dual) |
FEP |
ambient |
Wiegand [109] |
|
CSIRO Energy Technol Australia |
Indoor photoreactor black light |
18 |
FEP |
ambient |
Hynes et al. [110]; |
|
CSIRO Australia |
Indoor photoreactor black light |
~24 |
FEP |
ambient |
White et al. [52] |
|
CNRS-ICARE (HELIOS), France |
Outdoor |
90 |
FEP |
ambient |
Ren et al. [111] |
|
Nat’l Inst. Environ. Studies Japan |
Indoor photoreactor xenon arc |
6 |
PFA |
-- |
Akimoto et al. [62] |
|
Nat’l Inst. Environ. Studies Japan |
Indoor photoreactor
|
18 |
FEP |
ambient |
Sato et al. [112] |
|
Tsinghua University China |
Indoor photoreactor |
2 |
FEP |
283–333 |
Wu et al. [113] |
|
Key Lab. Organic Geochem. Guangzhou China |
Indoor photoreactor black light |
30 |
FEP |
283–313 |
Wang et al., [34] |
|
Key Lab. of Atmos. Inst. Comp. and Optical Radiation China |
Indoor photoreactor |
0.83 |
FEP |
298 |
Hu et al. [114] |
|
Kyungpook Natl Univ. Korea |
Indoor photoreactor black light |
7 |
FEP |
291–306 |
Babar et al. [35] |
|
Korea Institute of Science and Technol. |
Indoor photoreactor black light |
5.8 (dual) |
FEP |
ambient |
Bae et al.[31]; Moon et al. [115] |
a: References include example studies identified [37]; added references that describe the photoreactors from recent literature.
The photograph in Figure 2 is an example of another indoor facility at the Paul Scherrer Institute (PSI) in Switzerland. This chamber is part of cooperative research programs in Europe (e.g., EUROPHORE [95] or EUROCHAMP [116]). Special applications of chambers also have been reported, including portable bag experiments [117] to attempt to isolate the chemistry controlling oxidations of hydrocarbons, or characterize the reactivity of source emissions [118]. Exploration of in situ biogenic emissions from plants and interactions of plant VOCs with oxidant chemistry [119-121]. Other applications have used photochemical reaction products to investigate toxicity to humans [106,122]. Smog chambers have also been used to investigate photochemical pesticide degradation. Many pesticides contain chlorine, so that degradation can release this element to the O3 cycle [53, 123,124].
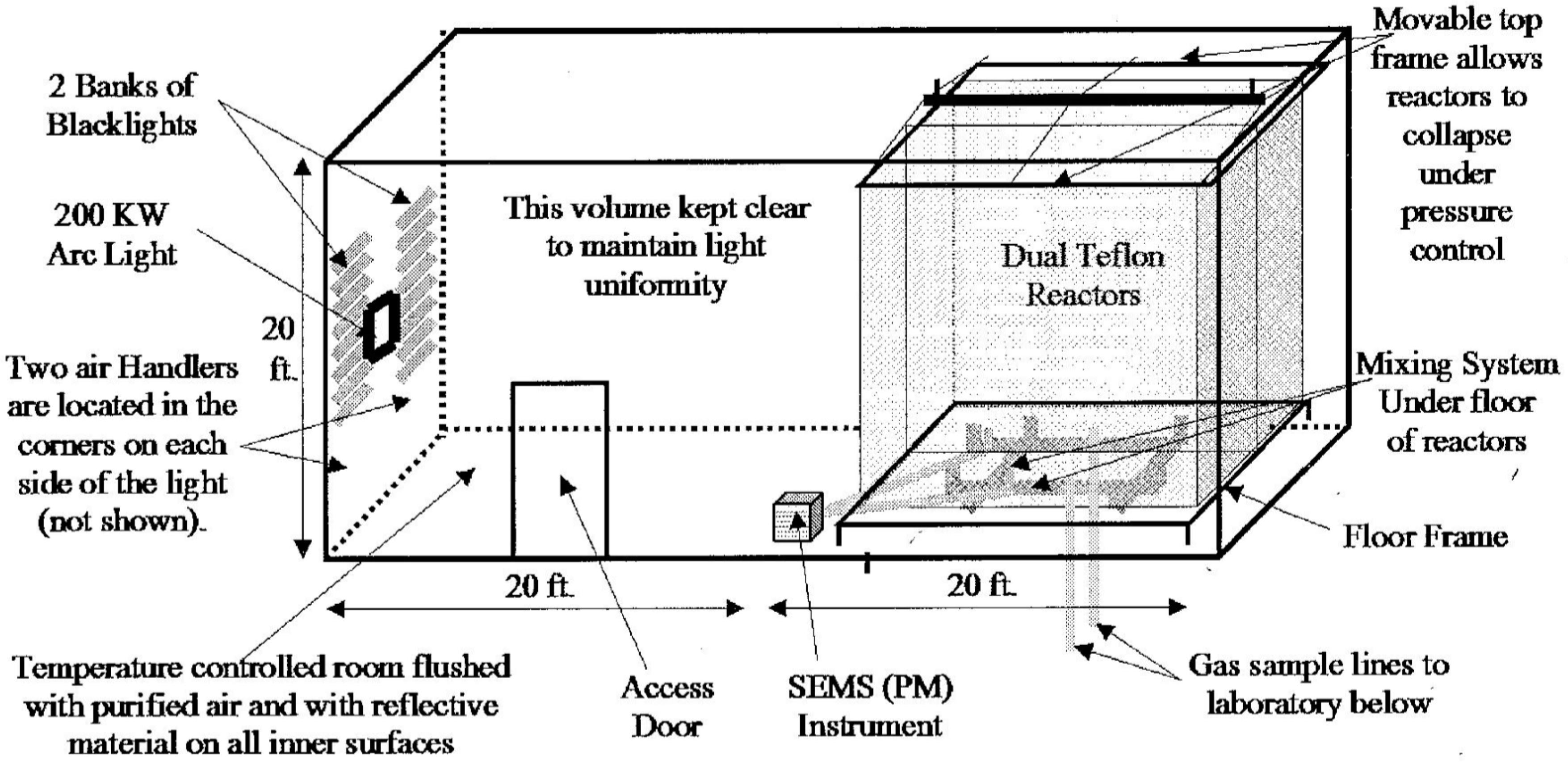
Figure 1. Example of Indoor Smog Chamber Configuration for the study of gas and aerosol photochemistry. CE-CERT, University of California Riverside System and the US Environmental Protection Agency (Reprinted from Carter et al., [85] with permission from Elsevier).

Figure 2. Photograph of the Indoor Smog Chamber at the Paul Scherrer Institute (Copyright Paul Scherrer Institute).
A second essential element for smog chamber research is the instruments for measurement of gases and aerosol particles. Measurements are sampled for external measurement or sampled in situ. Generic gas-phase experiments have relied on measurement of conventional O3, NO and NO2 concentrations and speciation for VOCs including hydrocarbons, oxygenates and organic nitrogen compounds (e.g., peroxyacetyl nitrate—PAN). Methods for measurement of these gases are described [28].
Smog chamber experiments provide a means for identifying precursor composition, intermediate species or organic products using different measurement techniques. These include gas chromatograph-mass spectrometer (GC-MS) characterization of VOCs, and instruments such as Fourier transform infrared spectroscopy (FTIR) or differential optical absorption spectrometry (DOAS). High-sensitivity options for VOC determination also include proton transfer spectrometry (PTS) [125]. Contemporary instruments for continuous monitoring include The Hantzsch instrument [126] for formaldehyde, and long path absorption photometer (LOPAP) for HONO [127]. Peroxide or hydroperoxide species can be determined using chemical ionization spectroscopy (CIMS) [128]. Use of high-performance liquid chromatography (HPLC) with detection using peridase [129] could also be adapted for hydroperoxide determination in smog chambers. Radical species such as OH and HO2 can be determined using fluorescence assay with gas expansion (FAGE) [130].
Aerosol experiments in chambers add complexity to measurement options. In addition to the gas-phase measurements, particle chemistry needs to be characterized, including physical properties such size distributions and chemical composition [131]. The range of particle size found in experiments (<0.01µm-~5µm diameter) has required the use of not only condensation nuclei counters (<~0.05 um), but also differential mass analyzers (0.05 µm-~5µm). Chemical composition of particles uses extracted samples obtained by filters for laboratory analysis, including inorganic and organic components by ion chromatography (IC), GC-MS, or aerosol mass spectrometry (AMS). Peroxides in particles have also been measured by LOPAP [132].
Organic species from reactants to condensed phase products require a variety of techniques including sampling for derivatization of species and subsequent laboratory analysis. Some of these adopt improvements in mass spectroscopy [133] including so-called “soft” ionization methods for large organic species (atmospheric pressure chemical ionization, API-MS [134], or electrostatic ionization, ESI- MS [135]). High-resolution time-of-flight mass spectrometers-HR T-of-F AMS have been applied to measure both gas-phase and condensed species atmospheric or smog chamber application (e.g, [136,137]). Gross et al. [138] used this instrument to measure continuously oligomers from SOA formation.
Quite apart from measurement capability, issues of sampling organic species from smog in recent years have been addressed It is widely known from ambient air sampling that reactive gas adsorption on tubing needs to be minimized and particles need to be sampled with attention to loss of volatile components of inorganic and organic species [139]. In the same way, sampling from chambers can bias measurements from tube wall losses. The wall loss from Teflon films applies also to various kinds of sample tubing, which in turn can bias low estimates of particle formation yields [140]. To facilitate combined sampling for gases and particles, a filter inlet (FIGAERO) has been adopted for mass spectrometric, chromatographic and other sensor applications. This sampler should make combined gas and particle characterization more efficient [136].
Aside from chemical kinetics evaluation, smog chambers have been used studies and documentation of instrument intercomparisons and interferences. Examples of these include NO2, HO2 and OH [141-143].
Limitations of Laboratory Chemical Simulation
There are obvious limitations to the smog chamber characterization of atmospherically relevant chemistry. The first are internal in nature, as discussed above, including simulation of sunlight from artificial light sources, completeness of mixing and chamber surface reactions. The second are external in nature concerning the coupling of chemical processes to other atmospheric phenomena. Internal limitations beyond surface reactions or reactant contamination are summarized next; external ambiguities are discussed below and later in Section 4. Finlayson-Pitts and Pitts [28] noted that the issue of wall effects is believed to be moot by some investigators. This is rationalized in that the relevance to the atmosphere is moderated by the fact that the atmosphere is adjacent to underlying surfaces. Photochemical processes along with aerosols take place with a wide range of surface area per unit volume of air near the ground (having widely varying properties).
Uncertainty introduced in translation from controlled laboratory experiments using defined or known gas mixtures vs. irradiation of urban or rural air is not necessarily well-established; experiments have elucidated this question with dual chambers or interpretation of smog evolution compared with oxidant forming mechanisms. In a sense, a “proof” of the relevance of chamber studies results from a correspondence with atmospheric concentrations and composition over durations including diurnal and longer term averages in urban or rural air (see also Section 4). The same heuristic argument is true for the influence of semi-continuous injections of reactants during experiments. The fact that, at least qualitatively, the diurnal changes in reactants and products are similar to those in the atmosphere is interpreted that this emission injection is accounted for in chemical transport models (see also Section 4).
Uncertainty in the translation of smog chamber experiments to current atmospheric conditions is also related to the latter ambient concentration levels. Emission control of precursor gases over the years has reduced urban concentrations substantially, and rural concentrations are generally much lower than current urban levels. Dodge [36], for example, noted this issue in 2000 and recommended experiments for low NOx and VOC concentrations and low VOC/NOx ratios paralleling contemporary conditions.
Another question of smog chamber experiments and the atmosphere is the presence of a “background” of photochemical reactants and products to begin each sunlight cycle. Most chamber experiments focus on highly purified air introduced with known pure VOC and NO gases, or in the case of aerosols pure salts or acids as seed species. In principle, the background issue could be dealt with to first order by conducting gas-phase experiments with a range of reactant concentrations and VOC/NOx ratios; this precludes an ability to simulate a background of partially reacted species, including free radicals, some of which may be unidentified. The latter may be moot, since these species in aged air are likely to be very short-lived and not relevant to a background residual of a day or more, some of which mixes down to the ground from the upper atmospheric boundary layer or free troposphere.
An illustration of the lifetimes expected for various VOC exposed to OH, NO3 radicals or O3 range from minutes to years (e.g., Table 1 from [38]), showing that background photochemistry of VOCs could be factor over multiple time scales. An early attempt to deal with the evolution of smog over a sequence of time scales to a few days was reported [50]. They used a dual outdoor chamber to simulate multiday degradation by a sequence of daily dilution in the chamber. Their results suggest a qualitative diurnal repetition of O3 forming reactions, but wall effects are likely to cumulatively affect the product concentrations.
The residence time for aerosols in the atmosphere varies from hours to several days depending on particle size and surface deposition rates or cloud or rainout [29]. For background perturbations associated with photochemical aerosol formation, the superposition of photochemical processes on known existing aerosol populations ranging from dust and carbonaceous material combined with inorganic salts poses a challenge for interpretation of the laboratory results to tropospheric aerosols. Nevertheless, the experiments using acid or metal oxide seeds simulates a real-world condition for catalyzing photochemical aerosol formation.
Another possible way to deal with background is to investigate the photochemical reactions of complex mixtures derived from combustion processes for comparison with the so-called “pure” systems. Chamber experiments of this kind have included reactions of internal combustion and diesel engine exhaust, as well as biomass burning [8, 77,144-152].
Smog chamber experiments generally address “dry” (RH< 70%) gas-phase or seed-catalyzed chemistry. These processes do not account for the potential of heterogeneous or condensed phase chemistry known to take place in the atmosphere. The role of clouds or fog for introducing uncertainty from light variation is exemplified by daily variation in sunlight for outdoor chambers. Perturbation in sunlight from cloud or haze intervention presumably acts to suppress photochemical aerosol formation and could be investigated as part of an experimental protocol. Reactions from microscale interactions with moist particles add complexity to irradiated chemical processing. Hallquist et al. [37] comment on cloud interactions that include the influence of RH on gas-particle partitioning, changes in water content with change in RH of droplets, or ice nucleation with organic content. This list does not address the potential for photochemical oxidant absorption and reaction with organics in droplets [153, 154]. Since the Hallquist et al. review, studies using humidity-controlled smog chambers and bulk laboratory methods have been reported [41,42]. Knowledge Gained from Chambers
Even with its limitations, chamber simulation of photochemical processes has contributed in many ways to advancing knowledge about the atmosphere. Smog chambers constructed since the 1950s have gone well beyond qualitative characterization of oxidant formation in the presence of VOC and NOx precursors. A large number of gas-phase kinetic simulations complementing other laboratory techniques have produced quantitative kinetic data for conditions near the atmosphere. Dodge [36] and Finlayson-Pitts and Pitts [28] have reviewed the kinetic and mechanistic studies published through the late 1990s. Smog chamber experiments have provided fertile ground for advancing knowledge of photochemical aerosol formation as well.
Atmospheric Photochemical Processes
A major contribution of smog chamber data for gas-phase processes involves their use in verification and evaluation of mechanisms relevant to the atmosphere. A large body of kinetic data for oxidation reactions was collected relating to VOCs and NOy species since the 1950s [38,155-157]. These have provided the ingredients constructing detailed mechanisms for oxidant production [158-162]. Potentially relevant reactions could involve more than 20,000 reaction pathways with thousands of trace species. Jenkin et al. [161] and Saunders et al. [162] constructed a gas-phase mechanism (Master Chemical Mechanism--MCM v3) involving NOy and 120 VOCs which translated to 7000 reactions with 2500 species.
Detailed mechanisms required too many computer resources for most atmospheric modeling applications. In practice, compressed or condensed mechanisms were adopted that evolved from highly parameterized schemes for O3 chemistry in the 1970s to more specific descriptions after the 1980s. Examples of compressed mechanisms include the SAPRC 07 (CS07A) model, or the RADM chemical models [162,163]. Both detailed and truncated mechanisms have been evaluated with smog chamber data. The detailed mechanism experiments added to the advanced understanding of the complex chemistry. Experiments with truncated mechanisms provided knowledge of the potential inaccuracies involved in constraining the oxidation chemistry.
In the US, three “streamlined” chemical models have been developed since the late 1970s. These are: (a) Carbon Bond (CB; [164-166]); (b) Regional Acid Deposition Model—RADM [163] (also the RADM derivative, Regional Atmospheric Chemistry Mechanism—ACM [167]); and (c) SAPRC series [158,159]. The Carbon Bond mechanism is perhaps most commonly used in the US regulatory environment, and has evolved through several modifications since their first versions; e.g., CB05 [166].
According to Dodge [36], the sequence of reactions involving NOy, O3, OH, HO2, H2O2 and CO is essentially the same except for the reaction of OH and NO2. The models adopt parameterized chemistry for VOC, particularly biogenic compounds and aromatics whose kinetic degradation mechanisms have been adjusted from smog chamber experiments to estimate O3 concentrations. Dodge’s Table 1 [36] lists over a thousand smog chamber runs exploring VOC-NOx systems that have been archived and have been used for guiding the fidelity of compressed model elements. Several hundred runs have been recorded and used to evaluate oxidant forming mechanisms since the 1970s.
An important conceptual outgrowth of application of chamber experiments that resulted in the NOx-VOC-O3 relationship traces back to Haagen-Smit’s research, and the reports of Romanovsky et al. [15] and Hamming et al. [14]. This relationship was represented from chamber experiments as the widely cited Haagen-Smit “mountain” diagram, shown, for example, in Figure 3. The diagram shows the two idealized regimes of initial NOx sensitivity and VOC sensitivity vs. the maximum O3 expected nominally during late morning to mid-day hours. The NOx sensitive regime is in the high initial VOC/NOx regime to the left of the line of slope of VOC/NOx ~8:1. The VOC sensitive regime of relatively low initial VOC/NOx is to the right of the ridge line. These regions are separated by isopleth contours that show maximum O3 formation with VOC/NOx ratio. The idealized relationship on Figure 3 was demonstrated early on from a selection of experiments with different VOC/NOx ratios. The relationship was calculated using the empirical kinetic modeling approach (EKMA) chemical model [168]. The mapping in the figure established a qualitative basis for precursor management that would estimate the average maximum expected O3 concentration. This approach evolved from simple, empirical (mainly VOC) roll-back schemes for O3 precursor reduction [169].
The current regulatory practices have evolved to adapt models for diurnal change in precursors using chemical transport models accounting for daily oxidant chemistry with precursor emissions and meteorological conditions for air transport and mixing [170]. Compressed kinetics calculations found in several air quality models, including the urban airshed (chemical transport) model (UAM) [171], the regional, RADM, “engineering” version [172] and versions of community multiple (spatial and temporal) scale chemical transport model (CMAQ) [173]. This portfolio of models is said to approximate by computer the “real” lower troposphere (see also Section 4).
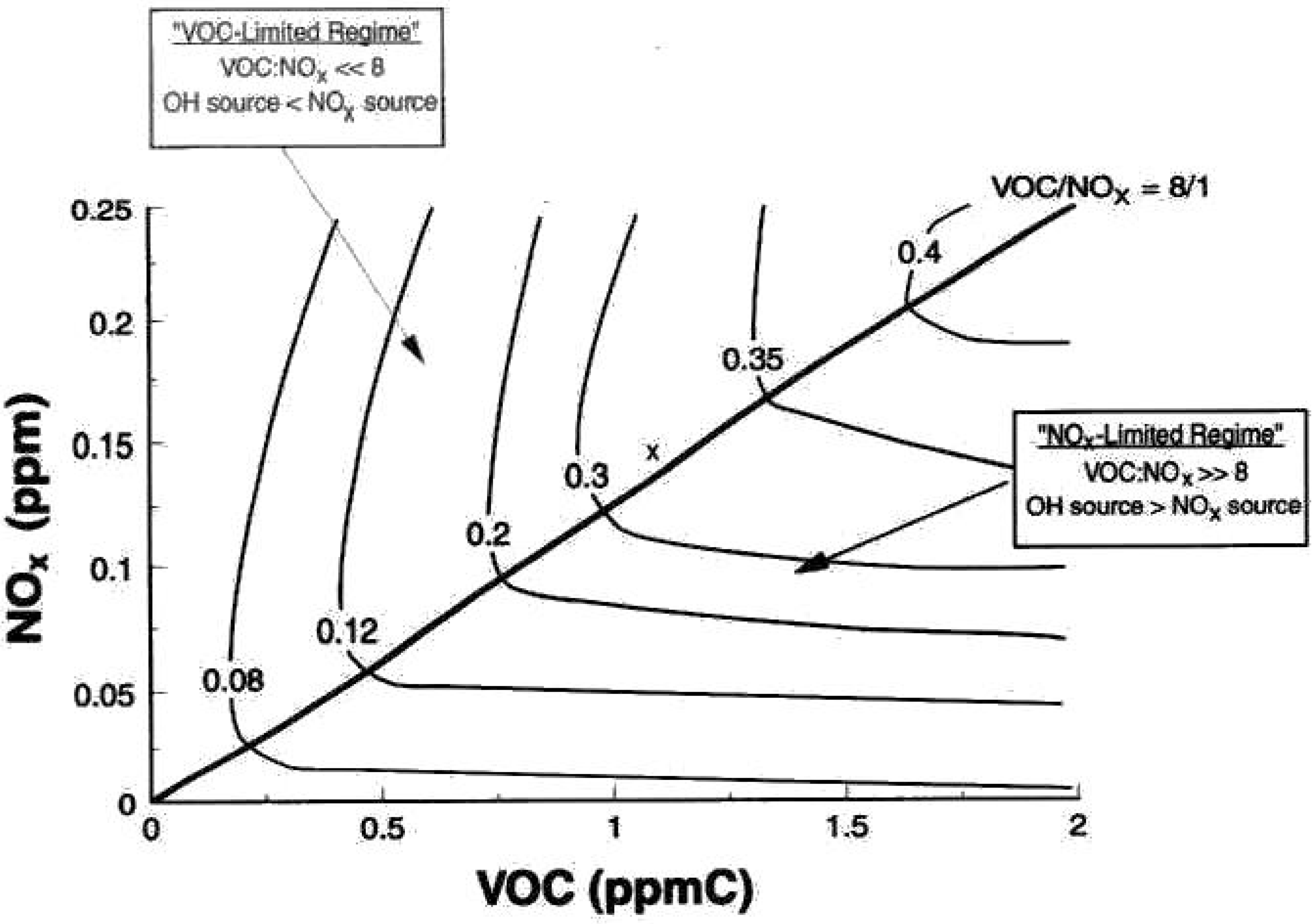
Figure 3. Textbook-handbook O3 Isopleth Diagram showing the non-linear relationship between O3 precursors and O3 concentrations as curved iso-lines calculated from the EKMA mechanism) [168].
Mechanism evaluation
Evaluation of photochemical mechanisms ideally would involve comparisons between the mechanistic calculations and smog chamber measurements of as many reactant and product species as practical for as long as possible. In most cases the evaluation is constrained for a given irradiation to concentration changes in a few hours for O3, NO, NO2, VOC species, products like formaldehyde or peroxyacetylnitrate (PAN) [174,175]. Recent evaluations can also address a variety of intermediate products including free radicals and peroxides.
A typical time series for smog chamber chemistry of a single aromatic compound over a period of a few hours is shown in the panels of Figure 4 with four different mechanism calculations. The upper panels follow O3, NO, NO2 and PAN; the lower panel shows the decline of o-xylene concentrations in the chamber. Here NO decreases exponentially after 0800 LDT, followed with a growth in NO2 concentration to a peak, and an O3 peak later in the run. Ozone then declined after ~1500 LDT. PAN concentration peaks at approximately noon.
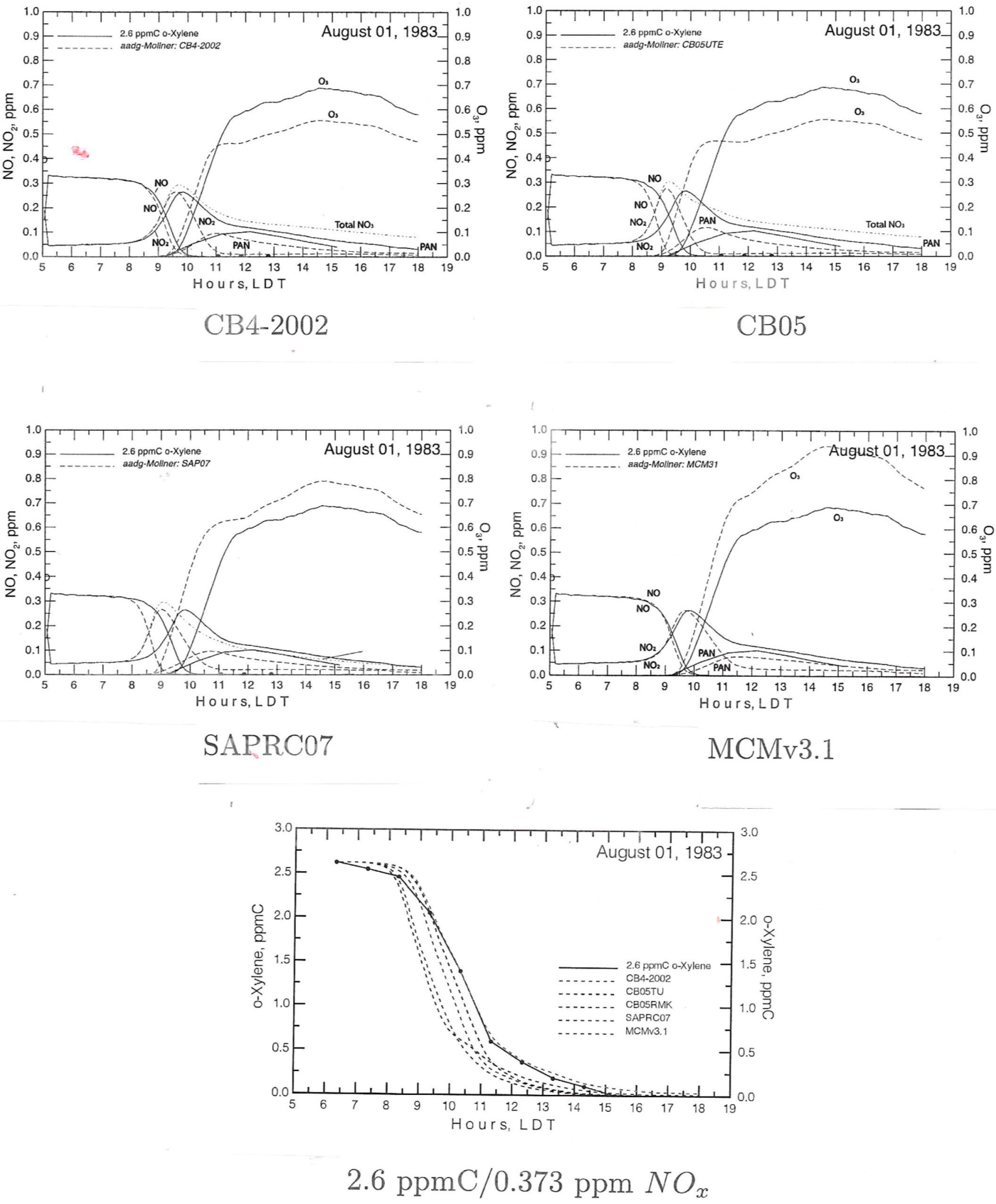 Figure 4. Smog chamber time series including a gas-phase reaction sequence of NOx and m-xylene, August 1, 1983, comparing data from the UNC outdoor chamber with CB04-2002, CB05, SAPRC-07 and MCMv3.1 calculations. The solid lines are the observations. Dotted lines are the model calculations (reprinted from [176], with permission from CSIRO publishing).
Figure 4. Smog chamber time series including a gas-phase reaction sequence of NOx and m-xylene, August 1, 1983, comparing data from the UNC outdoor chamber with CB04-2002, CB05, SAPRC-07 and MCMv3.1 calculations. The solid lines are the observations. Dotted lines are the model calculations (reprinted from [176], with permission from CSIRO publishing).
For comparison, the time variation from kinetics mechanisms is calculated and compared with the smog chamber runs. In this case, the CB mechanisms tend to under predict the O3 concentration, including the maximum and SAPRC and MCM over predict the measurements for O3; MCM is relatively poor for estimating the maximum O3 concentration in this experiment. The model predictions for NO and NO2 also show variable departure from the measurements. Comparison between the smog chamber observations and the different model chemistry has led to selective constraints on principal reaction pathways and rate constants. These results combined with similar testing with different chamber data facilitated improvements in mechanisms for simulating photochemistry in the atmosphere.
In another example, Carter and Lurmann [177] discuss a systematic evaluation of the SAPRC-90 detailed mechanism [158] with smog chamber data. One approach in their study is a test to see how well the mechanism predicts O3 concentrations for a range of different VOCs. Carter’s scatter plot in Figure 5 compares chamber measured O3 produced (O3-NO) after a period of time (5 h) with model calculated values [178]. The graph indicates the model calculation gives (O3-NO) production to within approximately 30% for a range of single VOC chamber experiments. This result is found even though the reactivity of the chosen VOCs varies widely.
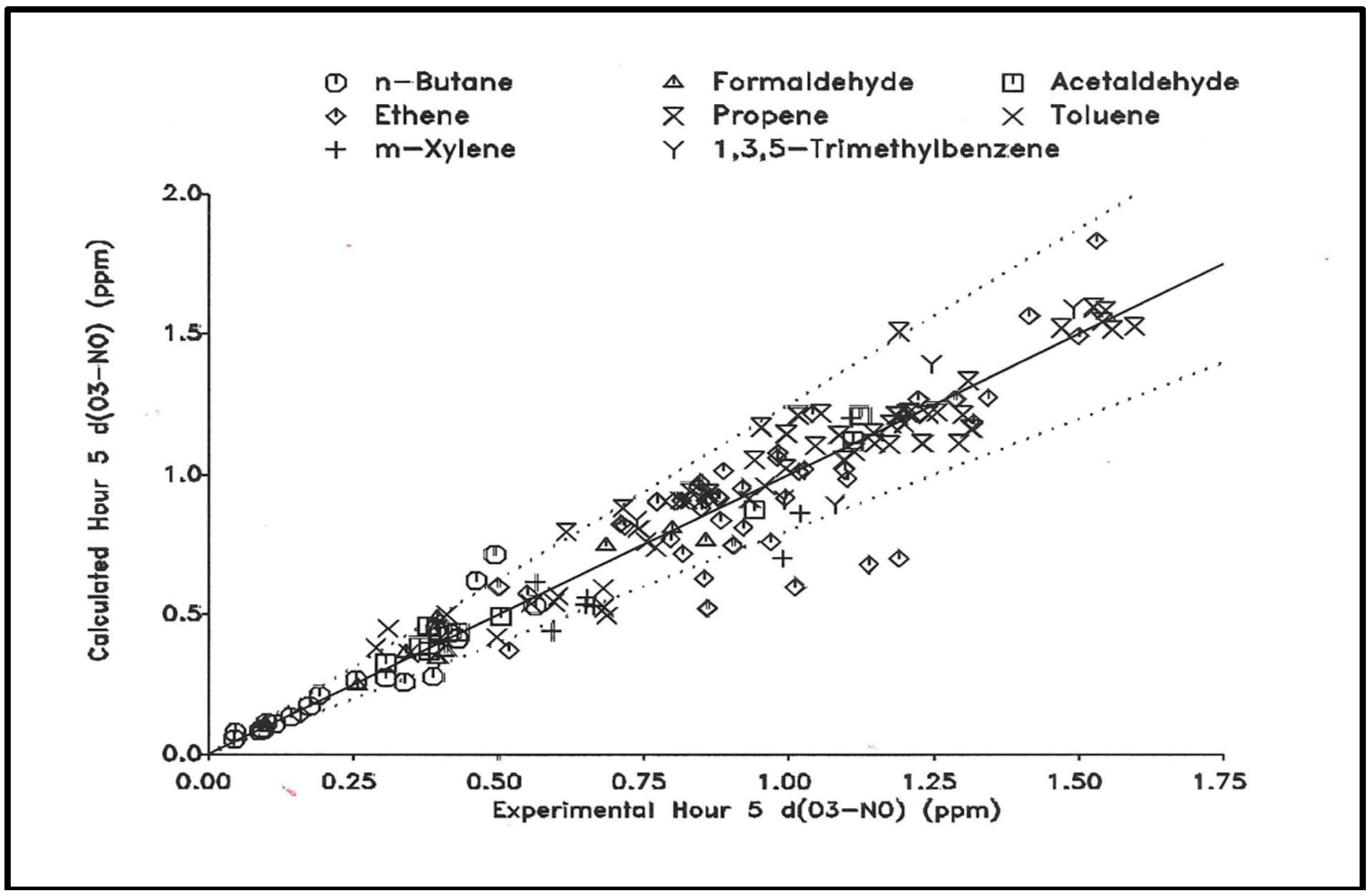
Figure 5. Experimental vs. calculated O3 formed plus NO oxidized for selected single compound-NOx experiments. Calculations were made using a version of the SAPRC-90 mechanism [178].
Relative Reactivity
An important result from smog chamber experiments concerns the influence of differences in reactivity of different VOC species. The reactivity of VOCs relates to the rate and amount of O3 or major oxidation products formed in the reaction sequence. VOC reactivity also affects reactions on overall free radical concentrations and the rate of NOy removal. Reactivity has been defined in different ways [178]. One simple measure is the reaction of OH on VOCs measured in terms of the OH radical rate constant (kOH). Another is the incremental reactivity, defined in terms of the change in O3 formation caused by the addition of a small amount of a VOC to the emissions in an O3 formation event; maximum incremental reactivity has been used as a useful index for mechanistic analyses [178].
There is a multitude of anthropogenic VOCs present in the atmosphere (e.g., alkenes, alkanes, aromatics and oxygenates). To characterize the influence of VOCs, initial practical approach was to combine species together using non- methane hydrocarbon (NMHC) concentration common to ambient air. The next step in complexity was to separate the VOC by the major ambient contributors by concentration. Then increased attention could be given to the reactivity of the individual species as potential contributors to the chemical process. The smog chamber experiments with various VOC species provided information about the capability of a mechanism to estimate reactivity. Then the correspondence of VOC reactivity differences in the atmosphere could be estimated from application of the mechanism in a chemical transport model.
Smog chamber experiments can give insight about the production of O3 from a variety of single VOCs as suggested in Figure 5. Alternatively, reactivity can be tested for a mixture by adding a VOC to the mixture and recording a change in O3 production. If adding a VOC to a mixture has sufficiently large effect on reducing free radical levels, then the addition may cause less O3 to be produced than in its absence. Conversely, if the VOC added increases radical levels then more O3 would be produced than in the VOC’s absence. Reactions of VOCs also increase rates of NOy removal, for example, by forming alkyl nitrates, inhibiting ozone production when NOy levels are low [179]. With NOy present, this VOC “sensitivity” is a practical consideration for managing atmospheric O3 production, especially for conditions of high VOC/NOx concentrations, or NOx sensitive conditions. When NOx concentrations are sufficiently low, O3 concentrations are very sensitive to NOx and less sensitive to VOC as reflected ideally in Figure 3.
Smog chamber experiments with NOx-VOC combinations are well-suited to test mechanisms used to calculate reactivity scales. Based on reactivity comparisons, scaling with incremental reactivity has been used to improve the SAPRC model results with experimental data in different gas-phase kinetic mechanisms [158, 159].
Photochemical Aerosols
In this section, the gas-phase smog chemistry is extended to include photochemical aerosol formation. Contemporary smog chamber experiments have followed the initial work between the 1950s and 1990. Chamber experiments have proven to be an important asset in developing knowledge about atmosphere relevant particle chemistry. They provide a means to: (a) establish how much particle mass is formed compared with reactants lost to gas-phase products and container walls (b) obtain yield data for parameterized SOA mechanisms for use in chemical transport or airshed models; (c) understand the link between gas-phase and particle chemistry, including physical and chemical properties of SOA; and (d) evaluate chemical mechanisms to predict SOA production rates for semi-volatile and low volatility species.
Recognition of aerosol formation from irradiated, atmospherically relevant gas mixtures in smog chamber experiments was noted after the 1950s as discussed earlier [8, 18,21]. These experiments inferred that there existed a clear linkage of oxidant chemistry and aerosol formation in smog instead of alternative sources such as smoke plumes. The experiments focused on sulfuric acid formation even though organics were present in the reacting mixture. Workers also recognized that particle nitrate could be formed through a pathway of nitric acid from the oxidant cycle [180]. Like their inorganic counterparts, organic aerosols are produced in the atmosphere as SOA. In the troposphere, high carbon number VOC precursors generally exist in the ppt-ppb level even in urban conditions.
Studies of the yield of photochemical reactions to produce low volatility material suggested that insufficient high carbon number vapors were present to explain major amounts of apparent photochemical SOA in smog, except for cyclic olefins and aromatics found in gasoline and other fuels. Grosjean and Friedlander [25] reported smog chamber measurements for some cyclic olefins that supported development of an aerosol-forming mechanism. Reactions analogous to those involving aromatics or cyclic VOC oxidize biogenic vapors. Isoprene, a relatively low-molecular-weight species, was thought to form products (low in carbon number) non-condensable at atmospheric concentrations. However, higher molecular weight species such as the terpenes (e.g., pinenes) could produce photochemically derived condensable products in ambient conditions where the reactants are present in concentrations less than ppb.
Smog chamber experiments have enabled the study of mechanisms relevant to photochemical aerosol--well beyond the early experiments of Los Angeles smog. Atmospheric chemical reactions of VOCs contribute to their degradation to final endpoints of CO and CO2. Intermediate stages of degradation by mass are mainly short-lived gas phase and condensed products. The photochemical formation of organic particles from carbon number species >C7 was established after the 1970s both in smog chamber experiments and in the field [23, 25,181–187]. The main thermodynamic criterion for aerosol formation was a low vapor pressure for products that would partition significantly into the condensed phase. The time series for particle production in chambers coupled with O3 as an index for oxidation; the increase in O3 concentration parallel with aerosol production are noted in comparison between the gas-phase product-time profiles in Figure 4. The relationship between O3 (or odd oxygen) and SOA concentrations also has been documented in ambient air, e.g., in the southeastern US [188] and in Mexico City [189].
A contemporary picture of the linkages between O3 photochemistry and photochemical aerosols is shown in Figure 6. For inorganic acid species, sulfuric acid and nitric acid, the photochemistry derives from oxidation via OH or H2O2 or O3. Secondary organic particle (SOA) formation follows from radical reactions or ozonolysis and depends on producing low vapor pressure (low volatility) species from higher volatility VOC vapors, partitioning semi-volatile product between the vapor and condensed phase. Reactions follow from an initial oxidation step generating products containing one or more oxygenated functional groups. Early on, one of the atmospheric pathways identified was an O3 attack on unsaturated VOC through formation of Criegee intermediates and ozonides [190] followed hypothetically by formation of oxygenated species of reduced vapor pressure [29]. Particle formation as a part of gas-phase oxidation reactions has been identified with OH, RO2, RO and NO3 radicals, and heterogeneous reactions on existing particle surfaces [29].
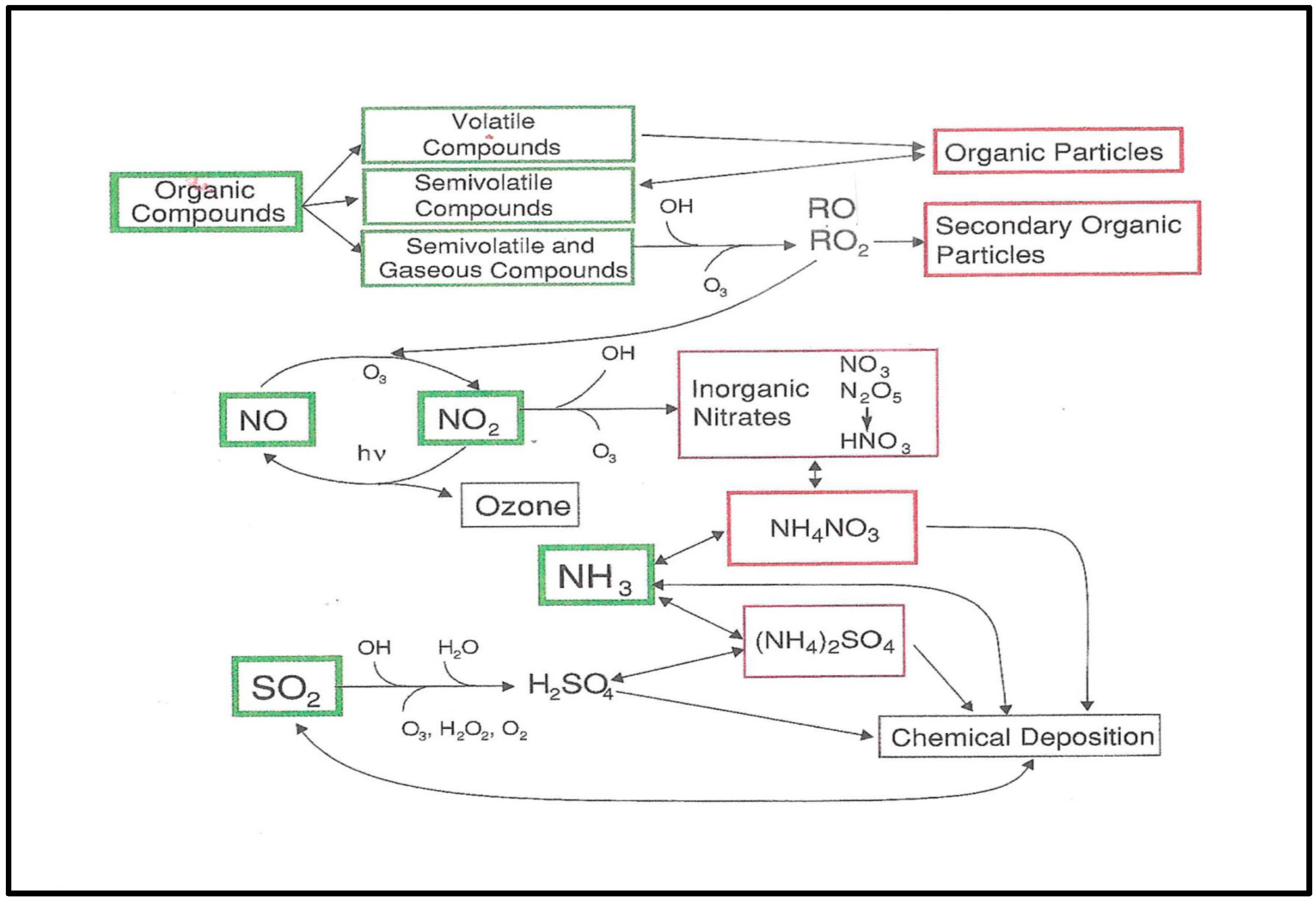
Figure 6. Interaction between the photochemical processing to form O3 and other oxidants through the VOC-NOx system combined with aerosol formation through SO2, NOy and VOC species. SOA chemical deposition is not included in the diagram (reproduced from [191], with permission of the Licencor through PLSclear).
Smog chamber and other reactor studies have indicated the presence of highly oxygenated material in photochemical aerosols than expected from oxidant attacks on large VOC molecules. As indicated in Table 3 addition of oxygenated functional groups to VOCs dramatically reduce vapor pressure, changing the partition between vapor and condensed phase. Vapor pressure reduction in smog chamber experiments has a direct effect on particle yields [192].
Table 3. Vapor pressure change of an organic compound with addition of a common functional group, based on group contributions calculated by Pankow and Asher [192]. (Reproduced from Kroll and Seinfeld [40] with permission from Elsevier).
|
Functional Group |
Structure |
Change in Vapor Pressure (298oK)a |
|
Ketone |
-C(O)- |
0.10 |
|
Aldehyde |
-C(O)H |
0.085 |
|
Hydroxyl |
-OH |
5.7 x 10-3 |
|
Hydroperoxyl |
-OOH |
2.5 x 10-3 |
|
Nitrate |
ONO2 |
6.8 x 10-3 |
|
Carboxylic acid |
-C(O)OH |
3.1 x 10-4 |
|
Peroxyacid |
-C(O)OOH |
3.2 x 10-3 |
|
Acyl peroxynitrate |
C(O)OONO2 |
2.7 x 10-3 |
|
Extra carbon |
-CH2- |
0.35b |
a: Multiplier with hydrocarbon root. b: For comparison between changes in polarity by addition of a functional group and changes in size of the carbon skeleton.
The yields of aerosol mass produced from photochemical reactions of VOCs (change in mass of aerosols/change in mass of VOC) relate to the amount of aerosol mass produced (Mo µg/m3). Yield curves for smog chamber reactions of C6-C9 species are illustrated in Figure 7. These curves are non-linear nature but indicate that the larger the yield or the larger the change in VOC, the larger the expected amount of aerosol relative to total VOCs present [193]. For single VOC species, the yield Mo relationship is qualitatively similar for many species, but the yield curves vary beyond simply a carbon number depending on the nature of the VOC vapor. At the turn of the 20th–21st century, two “discoveries” opened the door to an aggressive research program in photochemical aerosol formation. The first was the finding from smog chamber experiments that isoprene, a C5 unsaturated hydrocarbon, could form aerosols partly as a consequence of heterogeneous reactions with an acid catalyst [194,195] or other seed material [196-198]. “Seeding” may also be related to acid gases present in irradiated gas producing SOA [199]. The second finding derived in part from smog chamber and other experiments, indicated that aerosol “aging” in atmospheric conditions such that reactions modulated with a sequence of oxidation or cleavage reactions, and condensation to evaporation of semi-volatile species in successive chemical generations [40, 89, 200]
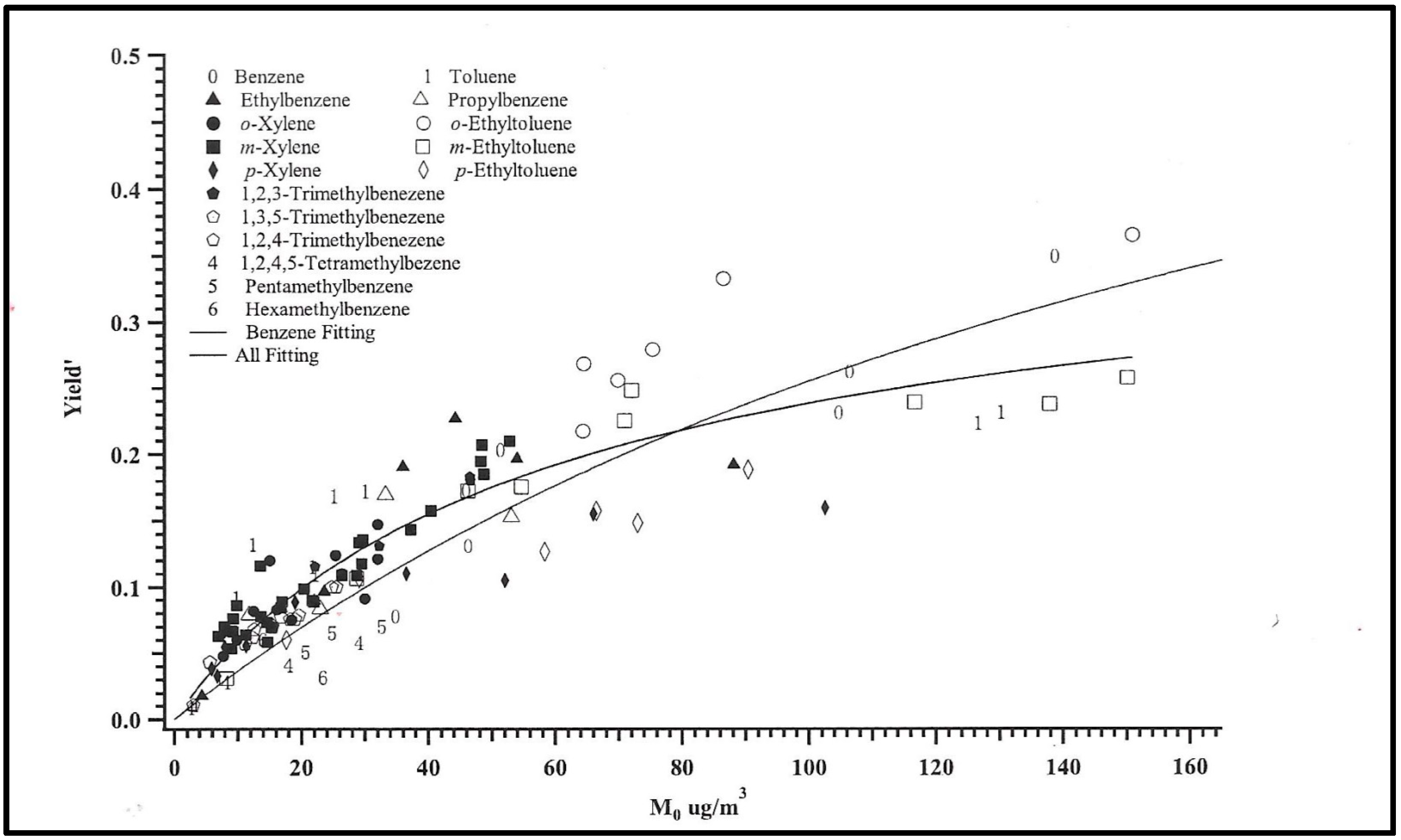
Figure 7. Yield curves for C6-C9 aromatic compounds from smog chamber experiments compared with VOC yield fitting. Yield is proportional to a mass-based stoichiometric coefficient and a partitioning coefficient. The shallower fitted yield curve at low Mo is that for benzene [193].
In the case of SOA formation from isoprene, Paulot et al. [201] and Surratt et al. [202] identified key intermediates in the oxidation process, including IEPOX (isoprene epoxydiols) and MPAN (methyl-acryloylperoxynitrate) that have also been found in the atmosphere. These intermediates play an important role in successive generation of oxygenated semi-volatile species from isoprene. They also open a pathway for organosulfate aerosol production from isoprene. Where isoprene is rich in VOC concentrations, organosulfate appears in the atmosphere as a fraction of the organic particles present [203].
Illustrated in Figure 8 are smog chamber results for aerosol production from isoprene oxidation in two NOx regimes. In both cases, particles measured by volume are formed after NO and isoprene are lost to the mixture. In the low NOx case, particle concentration reaches a maximum after 4 h with estimated >50% loss of isoprene, and weak O3 production of a few ppb. In the high NOx case, a substantial amount of O3 is produced, peaking at 3 h. Particle volume grows with O3, but levels at ~3 h. Kroll et al. [194] interpret the photochemical pathways in the NOx regimes in terms of the OH attack on the unsaturated carbon bond of isoprene producing degradation products from peroxy radical chemistry, including reduced volatility oxygenated species.
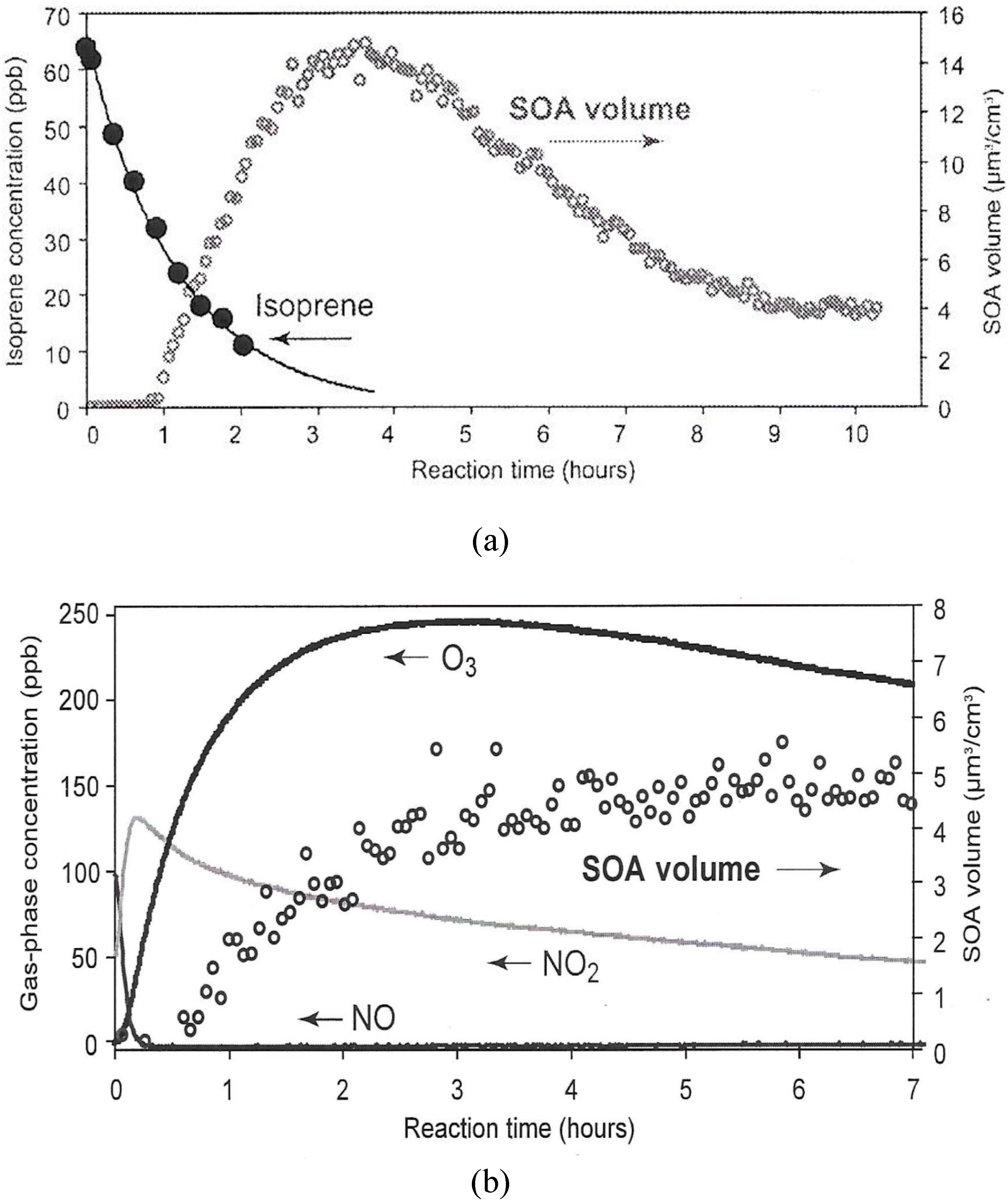
Figure 8. Examples of SOA production measured in terms of average particle volume change with irradiation time. Particle formation at low initial NOx concentration in air (< 1ppb), 63.6 ppb isoprene without inorganic seed (a) compared with high initial NOx concentrations (98 ppb NO; 31 ppb NO2); 42.7 ppb isoprene, and 6.4 µg/m3 (NH4)2SO4 seed (b) (Reprinted from [194] with permission from the American Chemical Society).
The SOA chemistry of isoprene and other VOCs involves oxidized products some of which lead to the formation of polymeric material, oligomers or humic-like substances [40,204], whose production pathways vary with NOx concentration. The aging of semi-volatile SOA is seen to involve a series of reaction “generations” with increasing oxygenation. “Aging” of semi-volatile VOCs by multiple generation chemical processes is potentially important on the order of a day timescale [83, 205,206].
Most kinetics experiments and derived mechanisms rely on the first generation of reactions and not subsequent processes. The evolution of particle composition during aging is hypothesized to involve organic species over a wide range molecular weight and oxygen content. The potential for interactions between high and low carbon number species created difficulty in seeking detailed mechanisms for aerosols to complement the gas-phase mechanisms. Two rules of thumb seem to hold for SOA from photochemical reactions--volatility and partitioning vapor pressure reduction, and increasing O/C ratio in particles with aging. The Donahue et al. [207] and Chacon-Madrid et al. [208] modeling approach embodies partitioning, and defines a volatility basis set (VBS) that involves saturation vapor concentration and the O/C ratio compared with the H/C ratio.
A third finding in recent years resurrects an important discussion of aqueous reactions to form sulfate [209]. As noted earlier, the oxidation of SO2 to form sulfuric acid is well known to involve aqueous reactions of OH, H2O2 and O3; Nitrate formation is influenced by equilibrium considerations involving HNO3, NH3 (or other cations) and H2O [210]. As noted above, heterogeneous processes on particles exist beyond catalyzed reactions [37]. The formation of SOA from aqueous reactions in the 1980s [153] has surfaced in recent studies. Evidence has emerged for photochemical (and non-photochemical) SOA production through aqueous, catalyzed reactions of water soluble organics, including glyoxal and hemiacetal, provided the medium for forming low-volatility compounds that are potentially embodied in wet, existing particles at high relative humidity, or clouds and fog [42, 211-213].
Laboratory study of aqueous reactions producing SOAs in the presence of hydrometeors is problematic for conventional smog chambers. However, smog chamber studies at varying humidities have added insight into SOA characterization, since generated particles are hygroscopic and contain major amounts of water at relative humidity above ~70% [ 214,215 ]. Reactions on wet aerosols or hydrometeors are mediated by organic surface coatings that complicate the liquid-gas interface [216,217].
Most photochemical aerosol models to date have relied on the application of smog chamber measurements for yield and a measure of VOC reduction, combined with thermodynamic estimates of phase partitioning. Formulation of chemical kinetic mechanisms for formation condensable species has lagged in development because of uncertainties in multi-generations of reaction pathways and products.
The assessment and interpretation of photochemical SOA production [218] guides researchers constructing kinetic models integrating gas-phase reactions and condensed phase processes. The aerosol dynamics involves not only chemical reactions, but also the physicochemical shaping the particle size-composition distribution. Kinetic modeling of aerosol dynamics traces back to the 1970s. For example, Middleton and Brock [219] reported a simulation that accounted for particle growth, coagulation, condensation-evaporation and nucleation, but without chemical production, per se. Examples of kinetic models for photochemical aerosol formation potentially applicable to atmospheric processes and supported with smog chamber data include those of [84,220-223]. Jathar et al. [224] have reported a three-dimensional kinetics model for use in air quality modeling. Current kinetic models used in CMAQ include CB05, RACM 2, SAPRC 07 and AERO-AE06 [225]. Vernecek et al. [226] reported the analysis of a preliminary version of the SAPRC-16 mechanism in CMAQ airshed modeling.
Continued progress to develop an aerosol kinetic model coupled with gas-phase chemistry called GECKO-A is an international collaborative enterprise [227,228]. This effort intends to synthesize available laboratory and field measurements with current kinetics mechanisms and models to produce a community-based model analogous to MCM. As part of this research, increased attention is likely to feed back to laboratory investigators that chamber studies are restricted to concentrations too high for relevance to contemporary ambient conditions. Autoxidation reactions [45,229], for example, may come into play in semi-volatile VOC oxidation to form highly oxidized molecules.
Evaluation of SOA models using smog chamber data follows from a history of evaluations of gas-phase mechanisms. Unlike the gas-phase mechanisms, however, SOA experiments can only rely on certain coarse comparisons. First, their reproducibility of results from chamber to chamber, and from semi-empirical model comparisons. Chamber experiments need to include not only the gas-phase measurements of temporal reactant and product change, but also aerosol properties such as nuclei concentration, mass or volume concentration and size distributions or AMS observations. Intercomparisons and evaluation studies have been reported that improve existing kinetics codes, for example [81,230,231].
The Atmosphere—Judging the Simulation Quality
As noted above, the gas-phase and aerosol mechanisms have been added to computer models to simulate the real atmosphere. The chemical components are tested for their relevance to atmospheric conditions in different ways. The relevance of gas-phase chemistry is well established through extensive and sustained measurements of ozone, its precursors and intermediate products, including the reactive nitrogen cycle and certain oxygenated organic compounds [28,43]. The presence of sulfate and nitrate and ammonium in atmospheric aerosols is also thoroughly documented in the literature [29, 232,233].
The relevance of photochemical SOA has relied on four different routes: (a) establishing the amount and temporal variation of SOA present relative to primary carbon sources, (b) measurement of tracer compounds that are expected from photochemical processes, (c) tracing daily time series of gases and particles across the earth’s surface, and (d) comparison of chemical transport (airshed) models with ground and aloft observations for gas-phase species, organic carbon and SOA.
The first goal establishes the apparent amount of SOA present relative to contributions from primary sources such as fuel combustion. The SOA variation by season would hypothetically associate photochemical aerosol presence with a seasonal maximum with O3. At least two methods exist for this determination. The first uses the difference between black and organic carbon ratios (OC/BC) from primary emissions (or OC/CO) vs. ambient values from which the secondary OC can be determined [234,188]. The second method derives from measurements using an AMS [235]. These observations have enabled the characterization of OC in terms of the ratios hydrocarbon rich (HOA) to reflect hydrocarbon- like species and oxygen rich organic aerosol (OOA) to identify with oxidized secondary species [236]. The OC/BC ratio methods and the AMS establish that SOA is a major component of OC in atmospheric aerosols, both in urban and rural-remote conditions.
The chemical tracer method provides direct evidence for SOA chemistry from identification of products from smog chamber studies, supporting their relevance to atmospheric processes. Tracer species for photochemical reactions, and their temporal variation represent key specific evidence of mechamisms. Organic tracers have identified characteristic molecular species with low volatility present in aerosols in special field campaigns. Species include certain oxygenated gases and condensed, or semi-volatile species such as the pinonic acid or pinonaldehyde from pinene oxidation, glyoxals, or isoprene epoxydiols or methyl tetrols from isoprene oxidation [108,237-239]. Organosulfates like C5H12O6S as intermediates also have been identified as tracers for biogenic SOA from smog chamber experiments [240,241]. While this approach has proven to be useful for some SOA chamber-based reactions, the method is constrained by sampling and analytical limitations for identifying and quantifying complex organic products expected in the first generation of oxidation and beyond [242].
As an alternative, the SOA contribution can be estimated from the difference between particles from primary OC sources and ambient species concentrations. Zheng et al. [243] report an example of this approach. Their analysis of urban conditions in the southeastern US identifies wood burning, transportation and meat cooking (and other, including SOA) to account for ~ 89% of the fine particle mass concentration, but the amount attributed to SOA and unknown sources was not resolved.
The third approach involves measurements of photochemical indicators in time and space in comparison with temporal measurements in smog chambers. These comparisons have been done for several modeling schemes. Conceptually the Lagrangian approach offers an analogy with smog chamber experiments. The USEPA developed a design for Lagrangian studies in the 1970s [244]. An experimental design was conceived in the 1973 Los Angeles Reactive Pollutant Program (LARPP), which used instrumented helicopters following the motion of tetroons for air mass tracking. Feigley et al. [245,246] conducted smog chamber experiments that were aimed at creating a comparative basis for measurements of O3, NOx and VOCs in the study conducted over Los Angeles in late summer and fall of 1973.
A similar Lagrangian-like experiment was conducted about the same time in Los Angeles [247]. This experiment involved the use of a blimp platform for O3, NO and NO2 instrumentation, and two indicators of particle production. The blimp was chosen for a capability to approximately mimic travel with an air mass across the Los Angeles Basin, which corresponds to a smog chamber run. The results of the flight are shown in Figure 9. The flight originated in relatively clean air near the Pacific Ocean; the blimp traveled in smog with the winds at a height of 150–300 m mslsl inland across the city. The portion of the flight that follows a track with air mass movement is between time 0 and 160 min.
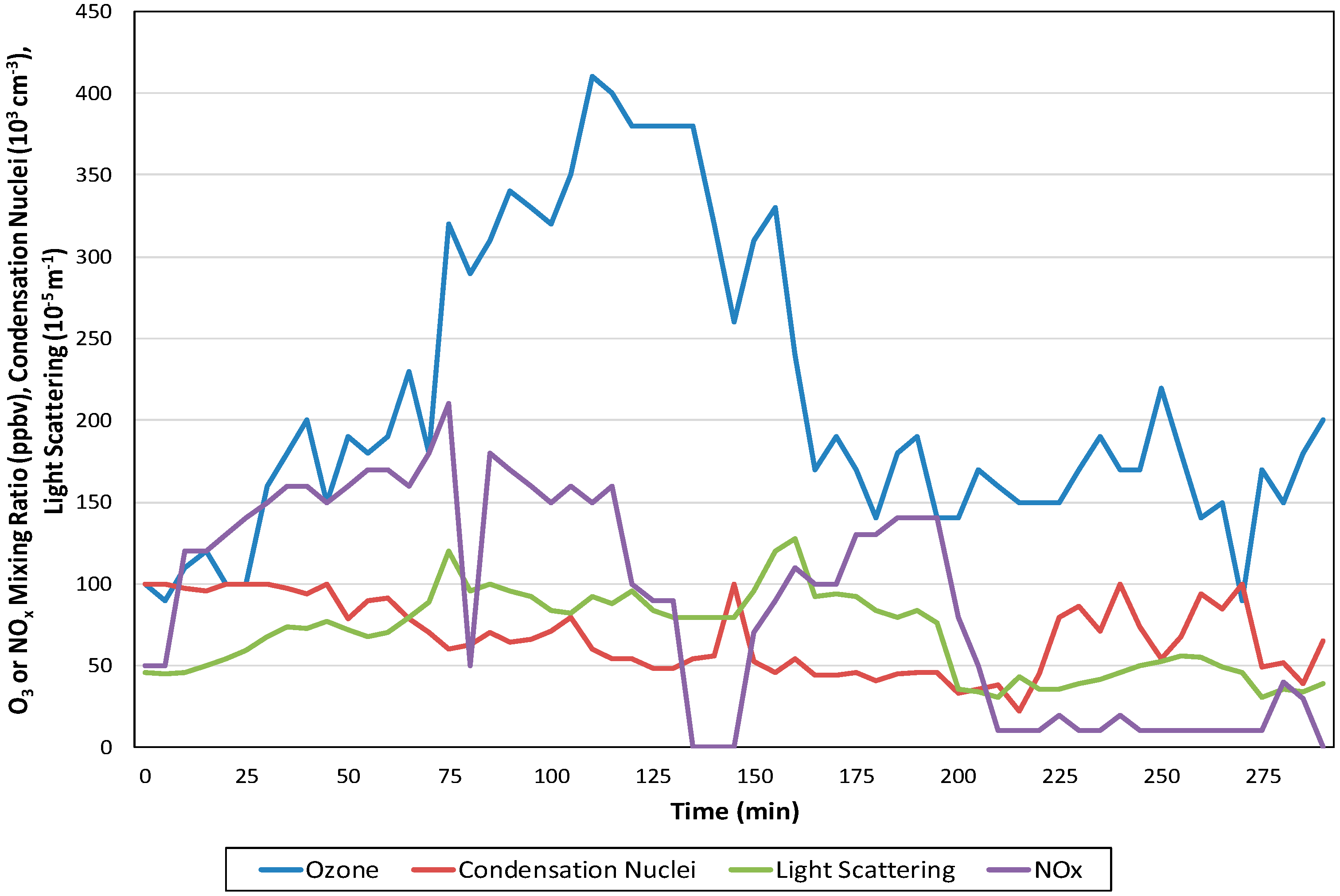
Figure 9. Lagrangian-like northwest to west trajectory observations from the blimp midday flight, September 6, 1973. The portion following air mass movement analogous to smog chamber O3 and particulate profiles is 0–165 min. The smog-chamber-like time series began and ended near Torrance, CA (US) [247] (reprinted with permission of the Air and Waste Management Association, www.awma.org).
The experiment showed the rapid rise of O3 and NOx early in the flight, followed by peaking of O3 mixing ratio after about 100 min. Both condensation nuclei concentrations--CN (~<0.05 µm diameter) and the particle light scattering coefficient (bsc) (mainly related to 0.1–5 µm diameter) depend on primary particle sources and secondary chemical production. The results of this experiment and parallel data for vertical concentration profiles demonstrate the complexity of interacting atmospheric processes over Los Angeles, far beyond the simulations in smog chambers. Notable among these are meteorological features involving the winds, thermal stratification, and transport differences with height [247,248].
The results of the blimp experiment indicated qualitatively the O3 production and NOx peaking took place as expected. In addition, evidence early in the experiment suggested link between light scattering and O3. The results support the presence of photochemical processing for both gases and a portion of particles. These in turn support the relevance of smog chamber experiments as simulators for atmospheric processes.
The fourth approach involves the application of chemical transport models as simulators embodying approximations of known atmospheric processes and the air–surface exchange (including emissions) occur continuously at various temporal and spatial scales [249]. The models most useful for mechanism relevance are those that have a time and space resolution of hours and a km or less. These contain gas-phase photochemical mechanisms and aerosol components that evolved mainly after the 1980s. The mechanisms are evaluated for fidelity of simulating the atmosphere with a combination of ambient measurements at the surface and aloft. The comparisons are supported with determination of the veracity of emissions data and meteorological observations. The chemical transport modeling can be extended to global averages over time and space constraints relevant to long-term phenomena; these are not discussed here, but are accessed in other venues [250-252].
Measurements for model testing exist from long-term averages from air monitoring of O3, NOy, VOCs, and particle mass concentration and composition. In addition, there are data from several experiments including major efforts characterizing O3 photochemistry [253] and sulfate and nitrate [254,255]. Particulate carbon in the presence of reactive gases also has been studied extensively in various projects [256-258].
Model performance compared with the observations as a reference has been reported as part of evolving model development and applications. Ideally comparisons are most useful for models with temporal resolution of an hour or less and spatial resolution of approximately a kilometer. Examples of deterministic model testing with photochemical mechanisms include those reported for CMAQ [259-263]. As an example for a 2002 Canadian exercise using a model similar to RADM [170], annual average O3 concentrations model estimates are 15–35% of observations. Typically, for average atmospheric sulfate, models are within 27–50%; performance is worse for nitrate 44%, and 85% for organic carbon. This difference between gas comparisons reflects in part the extensive experience with gas-phase models compared with other photochemical species. Other analyses have suggested that the volatility-based models underestimate measured SOA concentrations [40, 260]. Another example, a study of Volkamer et al. [264] in Mexico City, showed that reactive VOCs from anthropogenic sources produced much greater amounts of SOA than calculated from a volatility-based model.
Results of the kind noted indicate that the gas-phase and inorganic aerosol mechanisms adopted for chemical transport models support the photochemistry derived in part from smog chambers. For SOA, the aerosol chemistry in models needs additional work to give results expected from ambient measurements. The model and field campaigns provide a stimulus for continuing research on photochemical aerosols through linkages with smog chamber and other operations in collaborative programs like FIXCIT [265] and GECKO-A [227].
References
- Hidy,M. Adventures in Atmospheric Simulation. Bull. Am. Meteorol. Soc. 1967, 48, 143–161.
- Tyndall, J. On a new series of chemical reaction products by light. R. Soc. Lond. 1868, 17, 92–102.
- Wilson, C.T.R.; Grimes, R.W.; Catlow, C.R.A. Condensation of Water Vapour in the Presence of Dust-Free Air and Other Gases. Trans. R. Soc. A Math. Phys. Eng. Sci. 1897, 189, 265–307.
- Magill, P. Techniques Employed in the Analysis of Los Angeles Smog. In Proceedings of the First National Air Pollution Symposium, Los Angeles, CA, USA, 10–11 November 1949; pp. 61–68.
- Haagen-Smit, A.; Fox, M. Ozone formation in Photochemical Oxidation of Organic Substances. Eng. Chem. 1956, 48, 1484–1487.
- Renzetti, N. Ozone in Los Angeles Atmosphere. In Ozone Chemistry and Technology. Chem. 1959, 21, 230–282.
- Kehoe, R. Air Pollution and Community Health. In Proceedings of the First National Air Pollution Symposium, Los Angeles, CA, USA, 10–11 November 1949; pp. 115–120.
- Renzetti, N.; Doyle, G. The chemical nature of particulate in irradiated automobile exhaust. Air Pollut. Control Assoc. 1959, 8, 293–296.
- Stephens, E.; Schuck, E. Air pollution effects of irradiated auto exhaust as related to fuel consumption. Eng. Prog. 1958, 54, 71–77.
- Schuck, E.; Doyle, G. Photo-Oxidation of Hydrocarbons in Mixtures Containing Oxides of Nitrogen and Sulfur Dioxide; Report 29; Air Pollution Foundation: Los Angeles, CA, USA, 1959.
- Mader, P.; MacPhee, R.; Lafberg, R.; Larson, C. Composition of the organic part of atmospheric aerosols in Los Angeles. Eng. Chem. 1952, 44, 1352–1355.
- Mailila, J. On the early studies recognizing the role of sulfuric acid atmospheric haze and new particle formation. Tellus 2018, 70, 1–11.
- Richards, L. The Formation of Ozone in Polluted Atmospheres. Air Pollut. Control Assoc. 1956, 5, 216–246.
- Hamming, W.; Dickinson, J. Control of Photochemical Smog by Alteration of Initial Reactant Ratios. Air Pollut. Control Assoc. 1966, 16, 317–323.
- Romanovksy, J.C.; Ingels, R.; Gordon, R. Estimation of Smog Effects in the Hydrocarbon-Nitric Oxide System. Air Pollut. Control Assoc. 1967, 17, 454–459.
- Dimitriades, B. EPA Report 600/3-76-029; US Environmental Protection Agency: Research Triangle Park, NC, USA, 2005.
- Dimitriades, B. Methodology in Air Pollution Studies Using Irradiation Chambers. Air Pollut. Control Assoc. 1967, 17, 460–466.
- Harkins, J.; Nicksic, S. Studies of the Role of Sulfur Dioxide in Visibility Reduction. Air Poll. 1965, 15, 218–221.
- Stephens, E.; Price, M. Comparison of Synthetic and Smog Aerosols. Colloid Interface Sci. 1972, 39, 272–286.
- Wilson, W.; Merryman, E.; Levy, A.; Taliaferro, H. Aerosol Formation in Photochemical Smog. Air Pollut. Control Assoc. 1971, 21, doi.10.1080/00022470.1971.10469508.
- Wilson, W.; Levy, A.; Wimmer,A Study of Sulfur Dioxide in Photochemical Smog. J. Air Pollut. Control Assoc. 1972, 22, 27–32.
- Kocmond, W.; Yang, J.; Kittleson, S.; Whitby, K.; Demerjian, K. Aerosol Formation in Simple Photochemical Systems. In Fate of Pollutants in the Air and Water Environments; Part 2: Chemical and Biological Fate of Pollutants in Environment; Suffet, I., Ed.; Wiley-Interscience: New York, NY, USA, 1977; pp. 101–134.
- O’Brien, R.; Holmes, J.; Bockian, A. Formation of Photochemical Aerosols from Hydrocarbons. Sci. Technol. 1975, 9, 568–576.
- Heisler, S.; Friedlander, S. Gas-to-particle conversion in photochemical smog: Aerosol growth laws and mechanisms for organics. Environ. 1977, 11, 157–168.
- Grosjean, D.; Friedlander, S. Formation of Organic Aerosols from Cyclic Olefins and Diolefins. In The Character and Origins of Smog Aerosols; Hidy, G., Mueller, P., Grosjean, D., Appel, B., Wesolowski, J., Eds.; Wiley-Interscience: New York, NY, USA, 1980; pp. 435–476.
- Leighton, P. Photo Chemistry of Air Pollution; Academic Press: New York, NY, USA, 1961.
- A Theory of Upper-Atmospheric Ozone. Available Online: https://www.rmets.org/sites/default/files/papers/chapman-memoirs.pdf. (accessed on 10 July 2019)
- Finlayson-Pitts, B.; Pitts, J.N., Jr. Chemistry of the Upper and Lower Atmosphere; Academic Press: New York, NY, USA, 2000.
- Seinfeld, J.; Pandis, S. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change; Wiley-Interscience: New York, NY, USA, 2006.
- Paul Scherrer Institute. Smog Chamber Description. Available online: www.psich/lac/smog-chamber (accessed on 4 April 2019).
- Bae, G.; Kim, M.; Lee, S.; Song, K.; Jin, H.; Moon, K. Design and performance evaluation of the KIST indoor smog chamber. Korean Soc. Atmos. Environ. 2003, 19, 437–449.
- Becker, K. Overview on the Development of Chambers for the Study of Atmospheric Chemical Processes. In Environmental Simulation Chambers: Application to Atmospheric Chemical Processes; Barnes, I., Rudzinski, K., Eds.; Springer: New York, NY, USA, 2006; pp. 1–26.
- Wagner, R.; Bunz, H.; Linke, C.; Mohler, O.; Naumann, K.-H.; Saathoff, H.; Schnaiter, M.; Schurath, U. Chamber Simulations of Cloud Chemistry: The AIDA Chamber. In Environmental Simulation Chambers: Application to Atmospheric Chemical Processes; Barnes, I., Rudzinski, K., Eds.; Springer: New York, NY, USA, 2006; pp. 67–82.
- Wang, X.; Liu, T.; Bernard, F.; Ding, X.; Wen, S.; Zhang, Y.; He, Q.; Lu, S.; Chen, J.; Saunders, S.; et al. Design and characterizations of a smog chamber for studying gas-phase chemical mechanisms and aerosol formation. Meas. Tech. 2014, 7, 301–313.
- Babar, Z.; Park, J.-H.; Kang, J.; Lim, H.J. Characterization of a Smog Chamber for Studying Formation and Physicochemical Properties of Secondary Organic Aerosol. Aerosol Air Qual. Res. 2016, 16, 3102–2113.
- Dodge, M. Chemical oxidant mechanisms for air quality modeling: Critical review. Environ. 2000, 34, 2103–2130.
- Hallquist, M.; Wenger, J.C.; Baltensperger, U.; Rudich, Y.; Simpson, D.; Claeys, M.; Dommen, J.; Donahue, N.M.; George, C.; Goldstein, A.H.; et al. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Chem. Phys. Discuss. 2009, 9, 3555–3762.
- Atkinson, R. Atmospheric Chemistry of VOCs and NOx. Environ. 2000, 34, 2063-2101.
- George, C.; Ammann, M.; D’Anna, B.; Donaldson, D.; Nizkorodov, S. Heterogeneous Photochemistry in the Atmosphere. Rev. 2015, 115, 318–4258.
- Kroll, J.; Seinfeld, J. Chemistry of Secondary Organic Aerosol Formation and Evolution of Low Volatility Organics in the Atmosphere. Environ. 2008, 42, 3593–3624.
- Carlton, A.; Wiedinmyer, C.; Kroll, J. A review of secondary organic aerosol (SOA) formation from isoprene. Chem. Phys. 2009, 9, 4987–5005.
- Lim, Y.; Tan, Y.; Perri, M.; Serizinger, S.; Turpin, B. Aqueous chemistry and its role in secondary organic aerosol (SOA) formation. Chem. Phys. 2010, 10, 10521–10539.
- Stockwell, W.; Lawson, C.; Saunders, E.; Goliff, W. A Review of Tropospheric Chemistry and Gas-Phase Mechanisms for Air Quality Modeling. Atmosphere 2012, 3, 1–32.
- Glasius, M.; Goldstein, A. Recent Discoveries and Future Challenges in Atmospheric Organic Chemistry. Sci. Technol. 2016, 50, 2654–2764.
- Bianchi, F.; Kurten, T.; Riva, M.; Mohr, C.; Rissanen, M.; Roldin, P.; Berndt, T.; Crounse, J.; Wennberg, P. Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol. Rev. 2019, 119, 3472–3509.
- Lipeles, M.; Landis, D.; Hidy, G. The Formation of Organic Aerosols in a Fast Flow Reactor. In Fate of Pollutants in the Air and Water Environment, Part II; Suffet, I., Ed.; John Wiley and Sons: New York, NY, USA, 1977; pp. 69–94.
- Franzblau, E.; Burton, C.; Hidy, G. Aerosol Particle Formation from Ozone-Terminal Olefin Reactions. Aerosol Sci. Technol. 1984, 3, 167–176.
- Lambe, A.; Ahern, A.; Williams, L.; Slowik, J.; Wong, J.; Abbatt, J.; Brune, W.; Ng, N.; Wright, J.; Croasdale, D.; et al. Characterization of aerosol photooxidation reactors: Heterogeneous oxidation, secondary organic aerosol formation and cloud condensation nuclei activity measurements. Meas. Tech. 2011, 4, 445–461.
- Kang, E.; Root, M.; Toohey, D.; Brune, W. Introducing the concept of Potential Aerosol Mass (PAM). Chem. Phys. 2007, 7, 5727–5744.
- Sickles, J., II; Ripperton, L.; Eaton, W.; Wright, R. Oxidant-Precursor Relationships under Pollutant Transport Conditions; EPA Report 600/3-79-078a; US Environmental Protection Agency: Research Triangle Park, NC, USA, 1979.
- Jeffries, H.; Fox, D.; Kamens, D. Outdoor smog chamber studies: Light effects relative to indoor chambers. Sci. Technol. 1976, 10, 1006–1011.
- White, S.; Angove, D.; Kangwei, Li.; Campbell, I.; Element, A.; Halliburton, B.; Lavrencic, S.; Cameron, D.; James, I.; Azzi, M. Development of a new smog chamber for studying the impact of different uv lamps on SAPRC chemical mechanisms predictions and aerosol formation. Chem. 2018, 15, 171–182.
- Vera, T.; Munoz, A.; Rodenas, M.; Vazquez, M.; Mellouki, A.; Treacy, J.; Al Mulla, I.; Sidebottom, H. Photolysis of Trichloronitromethane (Chloropirin) under Atmospheric Conditions. Phys. Chem. 2010, 224, 1039–1057.
- Darnall, K.; Atkinson, R.; Winer, A.; Pitts, J., Jr. Effects of Constant vs. Diurnally-Varying Light Intensity on Ozone Formation. Air Pollut. Control Assoc. 1981, 31, 262–264.
- Dodge, M.; Bufalini, J.; Kopczynski, S. Contamination in Smog Chambers. Lett. 1972, 3, 101–109.
- Bufalini, J.; Theodore, A.; Marijon, M. Contamination Effects in O3 Formation in Smog Chambers. Sci. Technol. 1977, 11, 1181–1185.
- Lonneman, W.; Bufalini, J.; Kuntz, T.; Meeks, S. Contamination from Fluorocarbon Films. Sci. Technol. 1981, 15, 99–103.
- Kelly, N.; Olson, K.; Wong, C. Tests for Fluorocarbon and other Organic Vapor Release by Fluorocarbon Bags. Sci. Technol. 1985, 19, 361–364.
- Grosjean, D. Wall loss of Gaseous Pollutants in Outdoor Teflon Chambers. Sci. Technol. 1985, 19, 1059–1065.
- Yarwood, G.; Heo, G.; Carter, W.; Whitten, G. Environmental Chamber Experiments to Evaluate NOx Sinks and Recycling in Atmospheric Chemical Mechanisms; Report 06-2569981; Environ: Novaro, CA, USA, 2012.
- Trump, E.; Epstein, S.; Riipinen, I.; Donohue, N. Wall effects in smog chamber experiments: A model study. Aerosol Sci. Technol. 2016, 50, 1180–1200.
- Akimoto, H.; Hoshimo, M.; Inoue, G.; Sakamaki, F.; Washida, N.; Okuda, M. Design and Characterization of the Evacuable and Bakable Photochemical Smog Chamber. Sci. Technol. 1979, 13, 471–475.
- Carter, W.; Atkinson, E.; Winer, A.; Pitts, J., Jr. Evidence for Chamber-Dependent Radical Sources: Impact on Kinetic Computer Models for Air Pollution. J. Chem. Kinet. 1981, 13, 735–740.
- Carter, W.; Atkinson, R.; Winer, A.; Pitts, J., Jr. Experimental investigation of chamber-dependent radical sources. J. Chem. Kinet. 1982, 14, 1071–1103.
- Carter, W.; Atkinson, R.; Winer, A.; Pitts, J., Jr. The Wall as a Source of Hydroxl Radicals in Smog Chambers. Environ. 1985, 19, 1977–1978.
- Besemer, A.; Nieboer, H. The wall as a source of hydroxyl radicals in smog chambers. Environ. 1985, 19, 507–513.
- Sasamaki, F.; Akimoto, H. HONO Formation as Unknown Radical Source in Photochemical Smog Chamber. J. Chem. Kinet. 1988, 20, 111–116.
- Rohrer, F.; Bohn, B.; Brauers, T.; Bruning, D.; Johnen, F.-J.; Wahner, A.; Kleffmann, J. Characterisation of the photolytic HONO-source in the atmospheric simulation chamber SAPHIR. Chem. Phys. 2005, 5, 2189–2201.
- Iraci, L.; Johnston, J.; Golden, D. Experimental Techniques for Studying Surface Chemistry in Smog Chambers. Available online: www. cert.ucr.edu/-carter/epacham/iraci.pdf. (accessed on 10 April 2019).
- McMurry, P.; Rader, D. Aerosol losses in electrically charged chambers. Aerosol Sci. Technol. 1985, 4, 249–268.
- Charan, S.; Kong, W.; Flagan, R.; Seinfeld, J. Effect of particle charge on aerosol dynamics in Teflon environmental chambers. Aerosol Sci. Technol. 2018, 52, 854–871.
- McMurry, P.; Grosjean, D. Gas and Aerosol Wall Losses in Teflon Film Smog Chambers. Sci. Technol. 1985, 19, 1178–1182.
- Ye, P.; Ding, X.; Hakala, J.; Hofbauer, V.; Robinson, E.; Donahue, N. Vapor wall loss of semi-volatile organic compounds in a Teflon chamber. Aerosol Sci. Technol. 2016, 50, 822–9343.
- Matsunaga, A.; Ziemann, P. Gas-Wall Partitioning of Organic Compounds in a Teflon Film Chamber and Potential Effects on Reaction Product and Aerosol Yield Measurements. Aerosol Sci. Technol. 2010, 44, 881–892.
- Krechmer, J.; Pagonis, D.; Ziemann, P.; Jiminez, J. Quantification of Gas-Wall Partitioning in Teflon Environmental Chambers Using Rapid Bursts of Low Volatility Oxidized Species. Sci. Technol. 2016, 50, 5757–5765.
- Zhang, X.; Cappa, C.; Jathar, S.; McVay, R.; Ensberg, J.; Kleeman, M.; Seinfeld, J. Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol. Natl. Acad. Sci. USA 2014, 111, 5802–5807.
- Bian, Q.; May, A.; Kreidenweis, S.; Pierce, J. Investigation of particle and vapor wall-loss effects on controlled wood-smoke smog-chamber experiments. Chem. Phys. 2015, 15, 11027–11045, doi:10.5194/acp-15-11027-2015.
- Seinfeld, J. Effect of Vapor Loss in Laboratory Chambers on Yields of Secondary Organic Aerosols; Final Report to the California Air Resources Board; California Air Resources Board: Sacramento, CA, USA, 2015.
- Krechmer, J.; Day, D.; Ziemann, P.; Jiminez, J. Direct Measurements of Gas/Particle Partitioning and Mass Accomodation Coefficients in Environmental Chambers. Sci. Technol. 2017, 51, 11867–11875.
- Brune, W. The Chamber Wall Index for Gas-Wall Interactions in Atmospheric Environmental Enclosures. Sci. Technol. 2019, 53, 3645–3652.
- Pierce, J.; Englehart, G.; Hildebrandt, L.; Weitkamp, E.; Parthak, E.; Donahue, N. Constraining Particle Evolution from Wall Losses, Coagulation and Condensation-Evaporation in Smog Chamber Experiments: Optimal Based Size Distribution Measurements. Aerosol Sci. Technol. 2008, 42, 1001–1015.
- Sunol, A.; Charan, S.; Seinfeld, J. Computational simulation of the dynamics of secondary organic aerosol formation in an environmental chamber. Aerosol Sci. Technol. 2018, 52, 470–482.
- Karch, R.; Reischl, G.; Winkmayr, W.; Paulson, S.; Wang. S.; Yin, F.; Flagan, R.; Seinfeld, J. Smog Chamber Studies of Aerosol Formation in Atmospheric Mixtures. Aerosol Sci. 1989, 20, 995–998.
- Jathar, S.; Donahue, N.; Adams, P.; Robinson, A. Testing secondary organic aerosol models using smog chamber data for complex precursor mixtures: Influence of precursor volatility and molecular structure. Chem. Phys. 2014, 14, 5771–5780.
- Carter, W.; Cocker, D., III; Fitz, D.; Malkina, I.; Bumiller, K.; Sauer, C.; Pisano, J.; Bufalino, C.; Song, C. A new environmental chamber for evaluation of gas-phase chemical mechanisms and secondary aerosol formation. Environ. 2005, 39, 7768–7788.
- Cocker, D., III; Flagan, R.; Seinfeld, J. State of the art chamber facility for studying atmospheric aerosol chemistry. Sci. Technol. 2001, 35, 2594–2601.
- Ng, N.; Kwan, A.; Surratt, J.; Chan, A.; Chhabra, P.; Sorooshian, H.; Pye, J.; Crounse, P.; Wennberg, R.; Flagan, R.; et al. Secondary organic aerosol (SOA) formation from reaction of isoprene and nitrate radicals (NO3). Chem. Phys. 2008, 8, 4117–4140.
- Leone, J.; Flagan, R.; Grosjean, D.; Seinfeld, J. An Outdoor Smog Chamber and Modeling Study of Toluene-NOx Int. J. Chem. Kinet. 1985, 17, 177–216.
- Stanier, C.; Pathak, R.; Pandis, S. Measurements of volatility of aerosols from alpha-pinene ozonolysis. Sci. Technol. 2007, 41, 2756–2763.
- Robinson, A.; Donahue, N.; Shrivastava, M.; Weitkamp, E.; Sage, A.; Grieshop, A.; Lane, T.; Pierce, J.; Pandis, S. Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging. Science 2007, 315, 1259–1262.
- Mentel, T.; Bleilebens, D.; Wahner, A. A study of nighttime nitrogen oxidation oxidation in a large reaction chamber-the fate of NO2, N2O5, HNO3 and O3 at different humidities. Environ. 1996, 30, 4007–4020.
- Saathoff, H.; Naimann, K.-H.; Mohler, O.; Jonsson, A.; Hallquist, M.; Kiendler-Scharr, A. Temperature dependence of yields of secondary organic aerosols from the ozonolysis of a-pinene and limonene. Chem. Phys. 2009, 9, 1551–1577.
- Saathoff, H.; Moehler, O.; Schurath, O.; Kamm, S.; Dippel, B.; Mihekcic, L. The AIDA soot aerosol characterization campaign Aerosol Sci. 2003, 34, 1277–1296.
- Jonsson, A.; Hallquist, M.; Saathoff, H. Volatility of secondary aerosols from ozone-initiated oxidation of a-pinene and limonene. Aerosol Sci. 2007, 38, 843–852.
- Rodenas, M.; Munoz, A.; EUPHORE Team. The EUPHORE chambers: A shared research platform to study atmospheric processes-20 years of international cooperation. In Proceedings of the EGU General Assembly, Vienna, Austria, 23–28 April 2017.
- Klotz, B.; Sorenson, S.; Barnes, L.; Becker, K.; Etzkorn, T.; Volkammer, R.; Platt, U.; Wirtz, K.; Martin-Reviejo, M. Atmospheric oxidation of toluene in a large-volume outdoor photoreactor: In situ determination of ring-retaining product yields. Phys. Chem. 1998, 102, 10289–10299.
- Iinuma, Y.; Muller, C.; Berndt, T.; Boge, O.; Claeys, M.; Hermann, H. Evidence for the existence of organosulfates from beta-pinene ozonolysis in ambient secondary organic aerosol. Sci. Technol. 2007, 41, 6678–6683.
- Paulsen, D.; Dommen, J.; Kalberer, M.; Prevot, A.; Richter, R.; Sax, M.; Steinbacher, M.; Weingartner, E.; Baltensberger, U. Secondary Organic Aerosol Formation by Irradiation of 1,3,5-Trimethylbenzene-NOx-H2O in a New Reaction Chamber for Atmospheric Chemistry and Physics. Sci. Technol. 2005, 39, 668–2678.
- Temime, B.; Healy, R.; Wenger, J. A denuder-filter sampling technique for the detection of gas and particle phase carbonyl compounds. Sci. Technol. 2007, 41, 6514–6520.
- Healy, R.; Wenger, J.; Metzger, A.; Duplissy, J.; Kalberer, M.; Dommen, J. Gas/particle partitioning of carbonyls in the photooxidation of isoprene and 1, 3, 5-trimethylbenzene. Chem. Phys. 2008, 8, 3215–3220.
- Centre for Atmospheric Science. Smog Chamber Description. University of Manchester. Available online: www.manchester.ac.uk/restools/aerosolchamber/description (accessed on14 April 2019).
- Song, C.; Na, K.; Cocker, D, III. Impact of the Hydrocarbon to NOx Ratio on Secondary Organic Aerosol Formation. Sci. Technol. 2005, 39, 31143–33149.
- Tobias, M.; Ziemann, P. Kinetics of the gas-phase reactions of alcohols, aldehydes, carboxylic acids and water with C13 stabilized Criegee intermediate formed from ozonolysis of 1-tetradecene. Phys. Chem. 2001, 105, 6129-6135.
- Jeffries, H.; Fox, D.; Kamens, R. Outdoor Smog Chamber Studies (Effect of HC Reduction on Nitrogen Dioxide); EPA-650/3-75-011; Environmental Protection Agency: Research Triangle Park, NC, USA, 1975.
- Jeffries, H.; Kamens, R.; Sexton, K.; Gerhardt, A. Outdoor Smog Chamber Experiment to Test Photochemical Models; EPA Report 6003-82-016; Environmental Protection Agency: Research Triangle Park, NC, USA, 1982.
- Jeffries, H.; Kamens, R.; Sexton, K. Early history and rationale for outdoor chamber work at the University of North Carolina. Chem. 2012, 10, 349–364.
- Lee, S.; Jang, M.; Kamens, R. SOA formation from the photooxidation of a-pinene in the presence of freshly emitted diesel soot exhaust. Environ. 2004, 38, 2597–2605.
- Edney, E.; Kleindienst, T.; Jaoui, M.; Lewandowski, M.; Offenberg, J.; Wang, J.; Claeys, M. Formation of 2-methyl tetrols, and 2-methylglyceric acid in secondary aerosol from laboratory irradiated isoprene/NOx/SO2/air mixtures an their detection in ambient PM5 samples collected in the eastern United States. Atmos. Environ. 2005, 39, 5281–5289.
- Wiegand, A. Modelling Photochemical Production of Fine Particulates in a Toluene-NOx/Water Vapour System. Ph.D. Thesis, Queensland University of Technology, Brisbane, Australia, 1999.
- Hynes, R.; Angove, D.; Saunders, S.; Haved, H.; Azzi, M. Evaluation of two MCMv3.1 mechanisms using indoor environmental chamber data. Environ. 2005, 39, 7251–7262.
- Ren, Y.; Grosselin, B.; Daele, V.; Mellouki, A. Investigation of reaction of ozone with isoprene, methacrolein and methyl vinyl ketone using the HELIOS chamber. Faraday Discuss. 2017, 200, 289–311, doi:10.1039/c7fd00014f.
- Sato, K.; Hatakeyama, S.; Imamura, T. Secondary organic aerosol formation during photooxidation of toluene: NOx dependence on chemical composition. Phys. Chem. 2007, 111, 9796–9808.
- Wu, S.; Lu, Z.; Hao, J.; Zhao, Z.; Li, J.; Hideto, T.; Hiroaki, T.; Akio, Y. Construction and characterization of an atmospheric simulation smog chamber. Atmos. Sci. 2007, 24, 250–258.
- Hu, C.; Cheng, Y.; Pan, G.; Gai, Y.; Gu, X.; Zhao, W.; Wang, Z.; Zhang, W.; Chen, J.; Liu, F.; et al. A Smog Chamber Facility for Qualitative and Quantitative Study on Atmospheric Chemistry and Secondary Organic Aerosol. J. Chem. Phys. 2014, 6, 631–639, doi:10.1063/1674-0068/27/06/631-639.
- Moon, K.-C.; Bae, G.-N.; Lee, S.-B.; Choi, J.-E. Some Study Using Twin Chambers Filled with Ambient Air; Air Resources Research Center, Korea Institute of Science and Technology: Seoul, Korea, 2019.
- Doussin, T.; Formenti, P.; Gratien, A.; Cirtog, M.; Cazaunau, M.; Michoud, V.; Baltensberger, U.; Prati, P.; Brotto, P.; Munoz, A.; et al. Eurochamp 2020. Available online: www.researchgate.net/project/EUROCHAMP2020 (accessed on 12 June 2019).
- Kaltsonoudis, C. A portable dual-smog-chamber system for atmospheric aerosol field studies. Meas. Tech. 2019, 12, 2733–2743.
- Miracolo, M.; Hennigan, C.; Raajan, M.; Gordon, N.; Lipsky, E.; Presto, A.; Donahue, N.; Robinson, A. Secondary aerosol formation from photochemical aging of aircraft exhaust in a smog chamber. Chem. Phys. 2011, 11, 41356–41414.
- Delaria, E.; Vieira, J.; Cremieux, J.; Cohen, R. Measurements of NO and NO2 exchange between the atmosphere and Quecus agrifolia. Chem. Phys. 2008, 18, 14161–14173.
- Mentel, T.; Wildt, J.; Kiendler-Scharr, A.; Kleist, E.; Tillmann, R.; Dal Maso, M.; Fisseha, R.; Hohas, T.; Spahn, H.; Uerlings, R.; et al. Photochemical production of aerosols from real plant emissions. Chem. Phys. 2009, 9, 4387–4406.
- Price, D. Field and Smog Chamber Studies of Agricultural Emissions and Reaction Products. Master’s Thesis, Utah State University, Logan, UT, USA, 2010.
- Baltensperger, U.; Dommen, J.; Alfarra, M.; Duplissy, J.; Gaeggeler, K.; Metzger, A.; Facchini, C.; Decesari, S.; Finessi, E.; Reinnig, C. Combined Determination of the Chemical Composition and Health Effects of Secondary Aerosols: The Polysoa Project. Aerosol Med. Pulm. Drug Deliv. 2008, 21, 145–154.
- Woodrow, J.; Crosby, Seiber, J. Vapor-phase photochemistry of pesticides. Residue Rev. 1983, 85, 1215–1224.
- Borras, E.; Rodenas, M.; Vasquesz, M.; Vera, T.; Munoz, A. Particulate and gas-phase products from the atmospheric degradation of chlorphyifos and chlorpyrifos-oxon. Environ. 2015, 123A, 112-130.
- deGouw, J.; Warneke, C. Measurement of VOCs in the Earth’s Atmosphere using Proton Transfer Spectroscopy. Mass Spectr. Rev. 2007, 26, 223–257.
- Kelly, T.; Fortune, C. Continous Monitoring of Gaseous Formaldehyde using an Improved Fluorescence Approach. J. Environ. Anal. Chem. 1994, 54, 249–263.
- Keffmann, J.; Heland, J.; Kurtencach, R.; Lorzer, J.; Wiesen, P. A new instrument (LOPAP) for detection of nitrous acid (HONO). Sci. Poll. Res. 2001, 9, 48–54.
- Crounse, J.; McKinney, K.; Kwan, A.; Wennberg, P. Measurements of gas-phase hydroperoxides by chemical ionization mass spectroscopy. Chem. 2006, 78, 6726–6732.
- Lee, M.; Noone, B.; O’Sullivan, D.; Heikes, B. Method for the Collection and HPLC Analysis of Hydrogen peroxide and C1 and C2 Hydroperoxides in the Atmosphere. Atmos. Ocean. Technol. 1995, 12, 1060–1070.
- Hard, T.; George, L.; O’Brien, R.; FAGE Determination of Tropospheric OH and HO2. Atmos. Sci. 1995, 52, 3354–3372.
- Cocker, D., III; Whitlock, N.; Collins, D.; Wang, J.; Flagan, R.; Seinfeld, J. Instrumentation for state-of-the-art aerosol measurements in smog chambers. In Proceedings of the Combined US/German Ozone/Fine Particle Science and Environmental Chamber Workshop, Riverside, CA, USA, 4–6 October 1999.
- Mertes, P.; Pfaffenberger, L.; Dommen, J.; Kalberer, M.; Baltensperger, U. Development of a sensitive long path absorption photometer to quantify peroxides in aerosol particles (Peroxide-LOPAP). Meas. Tech. 2012, 5, 2339–2348.
- Baltensperger, U.; Chirico, R.; DeCarlo, P.; Dommen, J.; Gaeggeler, K.; Heringa, M.; Li, M.; Prevot, A.; Alfarra, M.; Gross, D.; et al. Recent developments in the mass spectroscopy of atmospheric aerosols. J. Mass Spectrom. 2010, 16, 389–395.
- Bruins, A. Mass Spectroscopy with ion Sources Operating at Atmospheric Pressure. Mass Spectrom. Rev. 1991, 10, 53–77.
- Fenn, J.; Mann, M.; Meng, C.; Wong, S.; Whitehouse, C. Electrostatic ionization for mass spectroscopy of large biomolecules. Science 1989, 246, 64–71.
- Lopez-Hilfiker, F.; Mohr, C.; Ehn, M.; Rubach, F.; Kleist, S.; Wildt, J.; Mentrel, T.; Lutz, A.; Hallquist, M.; Worsnop, D.; et al. A novel method for online analysis of gas and particle composition: Description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO). Meas. Tech. 2014, 7, 983–1001.
- Lee, B.; Lopez-Hilfiker, F.; Mohr, C.; Kurten, T.; Worsnop, D.; Thornton, J. An Iodide-Adduct High Resolution Time-of-Flight Chemical Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds. Sci. Technol. 2014, 48, 6309–6317.
- Gross, D.; Galli, M.; Kalberer, M.; Prevot, A.; Dommen, J.; Duplissy, J.; Gascho, A.; Mertzger, A.; Alfarra, M.; Gaeggeler, K.; et al. Real time measurement of oligomeric species in secondary organic aerosol with the aerosol time-of-flight spectrometer. Chem. 2006, 78, 2130–2137.
- Liu, X.; Deming, B.; Sat, S.; Palm, B.; Talukder, R.; Roberts, J.; Veres, P.; Krechmer, J.; Thornton, A. Effects of Gas-Wall Interactions on Measurements of Semivolatile Compounds and Small Polar Compounds. Atmos. Atmos. Meas. Tech. 2019, 12, 3137–3149, doi:10.5194/amt-2019-52.
- Pagonis, D.; Krechmer, J.; de Gouw, J.; Jiminez, J.; Ziermann, P. Effects of gas-wall partitioning in Teflon tubing and instrumentation on time-resolved measurements of gas-phase organic compounds. Meas. Tech. 2010, 10, 4687–4696.
- Fuchs, H.; Ball, S.; Bohn, B.; Brauers, T.; Cohen, R.; Dorn, H.-P.; Dube, W.; Fry, J.; Haseler, R.; Heitmann, U.; et al. Intercomparison of measurements of NO2 concentrations in the atmospheric simulation chamber SAPHIR during the NO3Comp campaign. Meas. Tech. 2010, 3, 21–37.
- Fuchs, H.; Novelli, A.; Rolleter, M.; Hofzumahaus, A.; Pfannestill, E.; Kessel, S.; Edtbauer, A.; Willams, J.; Michoud, V.; Dusnter, S.; et al. Comparison of OH reactivity measurements in the atmospheric simulation chamber SAPHIR. Meas. Tech. 2017, 10, 4023–4053.
- Onel, L.; Brennan, A.; Gianella, M.; Ronnie, G.; Lawry Aguilla, A.; Hancock, G.; Whalley, L.; Seakins, P.; Richie, G.; Heard, D. An intercomparison of HO2 measurements by fluorescence assay by gas expansion and cavity ring down spectroscopy within HIRAC (Highly Instrumented Reactor for Atmospheric Chemistry). Meas. Tech. 2017, 10, 4877–4894.
- Jeffries, H.; Sexton, K.; Yu, J. Atmospheric Photochemistry Studies of Pollutant Emissions from Transportation Vehicles Operating on Alternative Fuels; Final Report; National Renewable Energy Laboratory: Golden, CO, USA, 1998.
- Wiesen, P. Investigation of Real Car Exhaust in the EURPHORE Chamber; Combined US/German Ozone/Fine Particle Science and Environmental Chamber Workshop: Riverside, CA, USA, 1999.
- Geiger, H.; Kleffmenn, J.; Wiesen, P. Smog chamber studies on the influence of diesel exhaust on photosmog formation. Environ. 2002, 36, 1737–1747.
- Chirico, R.; De Carlo, P.; Heringa, M.; Tritscher, T.; Richter, A.; Pevot, A.; Dommen, J.; Weungartne, E.; G.Wehrle, G.; Gysel, M.; et al. Impact of After treatment Devices on Primary Emissions and Secondary Organic Aerosol Formation Potential from in-Use Diesel Vehicles: Results from Smog Chamber Experiments. Chem. Phys. 2010, 10, 11545–11463.
- Zielinska, B.; Samy, S.; Seagrave, J.; Mc Donald, J.; Wirtz, K.; Vazquez, M. Investigation of atmospheric transformations of diesel emissions in the European Photoreactor (EURPHORE). In Proceedings of the 5th Asian Aerosol Conference, Kaohsinung, Taiwan, 26–29 August 2007.
- Weitkamp, E.; Sage, A.; Pierce, J.; Donahue, N.; Robinson, A. Organic Aerosol Formation from Photochemical Oxidation of Diesel Exhaust in a Smog Chamber. Sci. Technol. 2007, 41, 6969–6975.
- Jathar, S.; Gordon, T.; Hennigan, C.; Pye, H.; Pouliot, G.; Adams, P.; Donahue, N.; Robinson, A. Unspeciated organic emissions from combustion sources and their influence on the secondary organic aerosol budget in the United States. Natl. Acad. Sci. USA 2014, 111, 10473–10478.
- Bruns, E.; Krapf, M.; Orasche, J.; Huang, Y.; Zimmeman, R.; Drinovec, L.; Mocnik, G.; El-Haddad, I.; Slowik, J.; Dommen, J.; et al. Characterization of primary and secondary wood combustion products generated from different burner loads. Chem. Phys. 2015, 15, 2825–2841.
- Tkacik, D.; Robinson, E.; Ahern, A.; Sale, R.; Stockwell, C.; Veres, P.; Simpson, I.; Meinardi, S.; Blake, D.; Yokelson, R.; et al. A dual-chamber for quantifying the effects of atmospheric perturbations on secondary organic aerosol formation from biomass burning emissions. Geophys. Res. Atmos. 2017, 122, 6043–6058, doi:10/1002/2016JD025784.
- Graedel, T.; Wechsler, C. Chemistry within aqueous atmospheric aerosols and raindrops. Geophys. 1981, 19, 505–539, doi:10.1029/RG–10i004p00505.
- George, I.; Abbatt, J. Heterogeneous oxidation of atmospheric aerosol particles by gas phase radicals. Chem. 2010, 2, 713–722.
- Atkinson, R.; Carter, W. Kinetics and mechanisms of the gas-phase reactions of ozone with organic compounds under atmospheric conditions. Rev. 1984, 84, 437–470.
- Calvert, J.; Orlando, J.; Stockwell, W.; Wallington, T. The Mechanisms of Reactions Influencing Atmospheric Ozone; Oxford University Press: New York, NY, USA, 2015.
- International Union of Pure and Applied Chemistry (IUPAC). Task Group on Atmospheric Chemical Kinetic Data Evaluation; Evaluated Data Sheets; International Union of Pure and Applied Chemistry: Research Triangle Park, NC, USA, 2019.
- Carter, W. A detailed mechanism for the gas-phase atmospheric reactions of organics. Environ. 1990, 24, 481–518.
- Carter, W. Development of the SAPRC-07 chemical mechanism. Environ. 2010, 44, 5324–5335
- Jenkin, M.; Saunders, D.; Wagner, V.; Pilling, M. Protocol for the development of the Master Chemical Mechanism, MCM v3, (Part B): Tropospheric degradation of aromatic volatile organic compounds. Chem. Phys. 2003, 3, 181–193.
- Saunders, S.; Jenkin, M.; Derwent, R.; Pilling, M. Protocol for the development of the Master Chemical Mechanism, MCM v.3 (Part A): Tropospheric degradation of non-aromatic organic compound. Chem. Phys. 2003, 3, 161–180.
- Kerr, J.A.; Calvert, J. Chemical Transformation Modules for Eulerian Acid Deposition Models: Volume 2 The Gas Phase Chemistry; EPA Report 600/S3-85/015; US Environmental Protection Agency: Research Triangle Park, NC, USA, 1985.
- Stockwell, W.; Middleton, P.; Chang, J. The Second Generation Regional Acid Deposition Model Chemical Mechanism for Regional Air Quality Modeling. Geophys. Res. 1990, 95, 16343–16367.
- Whitten, G.; Hogo, H.; Killus, J. The carbon-bond mechanism: A condensed kinetic mechanism for photochemical smog. Sci. Technol. 1980, 14, 690–700.
- Gery, M.W.; Whitten, G.Z.; Killus, J.P.; Dodge, M.C. A photochemical kinetics mechanism for urban and regional scale computer modeling. Geophys. Res. Space Phys. 1989, 94, 12925.
- Yarwood, G.; Rao, S.; Yocke, M.; Whitten, G. Updates to the Carbon Bond Chemical Mechanism: CB05; Final Report to US; RT-0400675; Environmental Protection Agency: Washington, DC, USA, 2005.
- Goliff, W.; Stockwell, W. The Regional Atmospheric Chemistry Mechanism, version 2; University of California: Davis, CA, USA, 2008.
- Dodge, M. Combined use of modeling techniques and smog chamber data to derive ozone-precursor relationships. In Proceedings of the International Conference on Photochemical Oxidant Pollution and Its Control, Raleigh, NC, USA, 12–17 September 1976. EPA Report 600/3-77-001b; US Environmental Protection Agency: Research Triangle Park, NC, USA, 1977; pp. 881–889.
- National Academy Sciences (US). The Relationship of Emissions to Ambient Air Quality. Report of the Coordinating Committee on Air Quality Studies; Committee of Public Works, US Senate: Washington, DC, USA, 1974.
- Seigneur, C.; Dennis, R. Atmospheric Modeling. In Technical Challenges of Multipollutant Air Quality Management; Hidy, G., Brook, J., Demerjian, K., Molina, L., Pennell, W., Scheffe, R., Eds.; Springer: New York, NY, USA, 2011; pp. 299–339.
- Reynolds, S.; Roth, P.; Seinfeld, J. Mathematical modeling of photochemical air pollution-I. Formulation of the model. Atmos. Environ. 1973, 7, 1033–1061.
- Chang, J.; Middleton, P.; Stockwell, W.; Walcek, C.; Pleim, J.; Lansford, H.; Binkowski, F.; Madronich, S.; Seaman, N.; Stauffer, D.; et al. The Regional Acid Deposition Model and Engineering Model. State-of-Science/Technology; Report 4; National Precipitation Assessment Program: Washington, DC, USA, 1991.
- Binkowski, F.; Roselle, S. Models 3-Community Multiscale Air Quality Model (CMAQ) model aerosol component 1. Model description. J. Geophys. Res. 2003, 108, doi:10.1029/2001/JD001409.
- Simonaitis, J.; Meagher, J.; Bailey, E. Evaluation of the condensed carbon bond (CB-IV) mechanism against smog chamber data at low VOC and NOx concentrations. Atmos. Environ. 1997, 31, 27–43.
- Bloss, C.; Wagner, V.; Bonzanini, A.; Jenkin, M.; Wirtz, K.; Martin-Reviejo, M.; Pilling, M. Evaluation of detailed aromatic mechanisms (MCMv3 and MCMv3.1) against environmental chamber data. Atmos. Chem. Phys. 2005, 5, 623–639.
- Parikh, M.; Jeffries, H.; Sexton, K.; Luecken, D.; Kamens, R.; Vizuete, W. Evaluation of Aromatic Oxidation Reactions in Seven Chemical Mechanisms with an Outdoor Chamber. Environ. Chem. 2013, 10, 245–259.
- Carter, W.; Lurmann, F. Evaluation of a detailed gas-phase atmospheric reaction mechanism using environmental chamber data. Atmos. Environ. 1991, 25, 2771-2806.
- Carter, W. Status of Research on VOC Reactivity in the United States. In Proceedings of the 1st US-German Workshop on the Photochemical Ozone Problem and its Control, Berlin, Germany, 24–27 September 1996.
- Farmer, D.; Perring, A.; Wooldridge, P.; Blake, D.; Baker, A.; Meinardi, S.; Huey, L.; Tanner, D.; Vargas, O.; Cohen, R. Impact of organic nitrates on urban ozone production. Atmos. Chem. Phys. 2011, 11, 4085–4094.
- Spicer, C.; Miller, D. Nitrogen Balance in Smog Chamber Studies. J. Air Pollut. Control Assoc. 1976, 26, 45–50.
- Hidy, G.M.; Burton, C.S. Atmospheric Aerosol Formation by Chemical Reactions. The Character and Origins of Smog Aerosols; Hidy, G.M., Mueller, P., Grosjean, D., Appel, B, Wesolowski, J., Eds.; Wiley-Interscience: New York, NY, USA, 1980; pp. 385–432.
- Grosjean, D.; Seinfeld, J. Parameterization of the Formation Potential of Secondary Organic Aerosols. Atmos. Environ. 1989, 23, 1733–1747.
- Odum, J.; Hofmann, T.; Bowman, F.; Collins, P.; Flagan, R.; Seinfeld, J. Gas Particle Partitioning and Secondary Organic Aerosol Yields. Environ. Sci. Technol. 1996, 30, 2580–2585.
- Ng, N.; Kroll, J.; Chan, A.; Chabra, P.; Flagan, R.; Seinfeld, J. Secondary aerosol formation from m-xylene, toluene and benzene. Atmos. Chem. Phys. 2007, 7, 3909–3922.
- Nolting, F.; Behnke, W.; Zetzsch, C. A smog chamber for studies of the reactions of terpenes and alkanes with ozone and OH. J. Atmos. Chem. 1988, 6, 47–59.
- Hu, D.; Tolocka, M.; Li, Q.; Kamens, R. A kinetic mechanism for predicting secondary organic aerosol formation from toluene oxidation in the presence of NOx and natural sunlight. Atmos. Environ. 2007, 41, 6478–6496.
- Matsunaga, A.; Docherty, K.; Lim, Y.; Ziemann, P. Composition and yields of secondary organic aerosol formed from OH radical-initiated reactions of linear alkenes in the presence of NOx: Modeling and measurements. Atmos. Environ. 2009, 43, 1348–1357.
- Blanchard, C.; Hidy, G.; Tanenbaum, S.; Edgerton, E.; Hartsell, B.; Jansen, J. Carbon in southeastern U.S. aerosol particles: Empirical estimates of secondary organic aerosol formation. Atmos. Environ. 2008, 42, 6710–6720.
- Herndon, S.; Onasch, T.; Wood, E.; Kroll, J.; Canagaratna, M.; Jayne, J.; Zavala, M.; Knighton, W.B.; Mazzoleni, C.; Dubey, M.; et al. Correlation of secondary organic aerosol with odd oxygen in Mexico City. Geophys. Res. Lett. 2008, 35, L15804.
- O’Neal, H.E.; Blumstein, C. A new mechanism for gas phase ozone-olefin reactions. Int. J. Chem. Kinet. 1973, 5, 397–413.
- McMurry, P., Shepherd, M., Vickery, J. (Eds.) Particulate Matter Science for Policy Makers; Cambridge University Press: Cambridge, UK, 2004; p. 22.
- Pankow, J.; Asher, W. SIMPOL.1: A simple contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds. Atmos. Chem. Phys. 2008, 8, 2773–2796.
- Cocker, D., III; Li, W.; Li, L.; Kacarab, M.; Peng, W. Air Quality Impacts of Low Vapor Pressure-Volatile Organic Compounds; Contract No. 13-302; Final Report; California Air Resources Board: Sacramento, CA, USA, 2016.
- Kroll, J.; Ng, N.; Murphy, S.; Flagan, R.; Seinfeld, J. Secondary organic aerosol formation from isoprene photooxidation. Environ. Sci. Technol. 2006, 49, 1869–1877.
- Jang, M.; Czoschke, N.M.; Lee, S.; Kamens, R.M. Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions. Science 2002, 298, 814–817.
- Hao, L.; Wang, Z.; Huang, M.; Fang, L.; Zhang, W. Effects of seed aerosols on the growth of secondary organic aerosols from the photooxidation of toluene. J. Environ. Sci. 2007, 19, 704–708.
- Chu, B.; Jiang, J.; Wang, K.; Li, J.; Hao, J. Effects of Inorganic Seeds on Secondary Organic Aerosol (SOA) Formation. In Atmospheric Aerosols-Regional Characteristics-Chemistry and Physics; IntechOpen: London, UK, 2012.
- Chu, B.; Liu, T.; Zhang, X.; Liu, Y.; Ma, Q.; Ma, J.; He, H.; Wang, X.; Li, J.; Hao, J. Secondary aerosol formation and oxidation capacity in photooxidation in the presence of Al2O3 seed particles and SO2. Sci. China Chem. 2015, 58, 1426–1434.
- Kleindienst, T.E.; Edney, E.O.; Lewandowski, M.; Offenberg, J.; Jaoui, M. Secondary Organic Carbon and Aerosol Yields from the Irradiations of Isoprene and a-Pinene in the Presence of NOx and SO2. l. Environ. Sci. Technol. 2006, 40, 3807–3812.
- Donahue, N.; Robinson, A.; Stanier, C.; Pandis, S. Coupled partitioning, dilution, and chemical aging of semi-volatile organics. Environ. Sci. Technol. 2006, 40, 2635–2443.
- Paulot, F.; Crounse, J.; Kjaergaard, H.; Kurten, A.; St. Clair, J.; Seinfeld, J.; Wennberg, P. Unexpected Epoxide Formation in the Gas-Phase Photooxidation of Isoprene. Science 2009, 325, 730–733.
- Surratt, J.; Chan, A.; Eddingsaas, N.; Chan, M.-N.; Loza, C.; Kwan, A.; Hersey, S.; Flagan, R.; Wennberg, P.; Seinfeld, J. Reactive Intermediates Revealed in Secondary Organic Aerosol Formation from Isoprene. Proc. Natl. Acad. Sci. USA 2010, 107, 6640–6645.
- Lopez-Hilfiker, F.; Mohr, C.; D’Ambro, E.; Lutz, A.; Riedel, T. Molecular Composition and Volatility of Organic Aerosol in the Southeastern U.S.: Implications for IEPOX Derived SOA. Environ. Sci. Technol. 2016, 50, 2200–2209.
- Kalberer, M.; Paulsen, D.; Sax, M.; Steinbacher, J.; Dommen, J.; Prevot, A.; Fisseha, R.; Weingartner, E.; Frankevich, V.; Zenobi, R.; et al. Identification of polymers as major components of atmospheric organic aerosols. Science 2004, 303, 1659–1662.
- Isaacman-Van Wertz, G.; Massoli, P.; O’Brien, R.; Lim, C.; Franklin, J.; Moss, J.; Hunter, J.; Nowak, J.; Canagaratna, M.; Misztal, P.; et al. Chemical evolution of atmospheric organic carbon over multiple generations of oxidation. Nat. Chem. 2018, 10, 462–468.
- Donahue, N.; Henry, K.; Mentel, T.; Kiendler-Scharr, A.; Spindler, C.; Bohn, B.; Brauers, T.; Dorn, H.; Fuchs, H.; Tillman, R.; et al. Aging of biogenic secondary organic aerosol for gas-phase radical reactions. Proc. Natl. Acad. Sci. USA 2012, 109, 13503–13508.
- Donahue, N.; Epstein, S.; Pandis, S.; Robinson, A. A two-dimensional volatility basis set: Organic-aerosol mixing thermodynamics. Atmos. Chem. Phys. 2011, 11, 3303–3318.
- Chacon-Madrid, H.; Murphy, B.; Pandis, S.; Donahue, N. Simulations of Smog-Chamber Experiments Using the Two-Dimensional Volatility Basis Set: Linear Oxygenated Precursors. Environ. Sci. Technol. 2012, 46, 11179–11186.
- Martin, L. Kinetic Studies of Sulfite Oxidation in Aqueous Solutions. Acid Precipitation: SO2, NO, NO2 Oxidation Mechanisms: Atmospheric Conditions; Calvert, J., Ed.; Ann Arbor Scientific Publications: Ann Arbor, MI, USA, 1984; pp. 63–100.
- Nenes, A.; Pandis, S.; Pilinis, C. ISORROPIA: A new thermodynamic model for multiphase multicomponent inorganic aerosols. Aquat. Geochem. 1998, 4, 123–152.
- Altieri, K.; Carlton, A.; Turpin, B.; Seitzinger, S. Formation of Oligomers in Cloud-Processing Reactions of Isoprene Oxidation Products. Environ. Sci. Technol. 2006, 40, 4956–4960.
- Ervens, B.; Turpin, B.; Weber, R. Secondary organic aerosol formation in cloud droplets and aqueous paricles (aqSOA): A review of laboratory, field and model studies. Atmos. Chem. Phys. 2011, 11, 11069–11102.
- Romonosky, D.; Li, Y.; Shiraiwa, M.; Laskin, A.; Laskin, J.; Nizkorodov, S. Aqueous Photochemistry of Secondary Aerosol of a-Pinene and a-Humulene Oxidized with Ozone, Hydroxyl Radical, and Nitrate Radical. J. Phys. Chem. 2017, 121, 1298–1309.
- Hinks, M.; Montoya-Aguilera, J.; Ellison, L.; Lin, P.; Laskin, A.; Laskin, J.; Shiraiwa, M.; Dabdub, D.; Nizkorodov, S. Effect of relative humidity on the composition of secondary organic aerosol formed from the oxidation of toluene. Atmos. Chem. Phys. 2018, 18, 1643–1652.
- Tillmann, R.; Hallquist, M.; Jonsson, A.; Kiendler-Scherr, A.; Saathoff, H.; Ianuma, Y.; Mentel, T. Influence of relative humidity and temperature on the production of pinonaldehyde and OH radicals from the ozonolysis of a-pinene. Atmos. Chem. Phys. 2010, 10, 7057–7072.
- Saxena, P.; Hildemann, L.M.; McMurry, P.; Seinfeld, J. Organics alter hygroscopic behavior of atmospheric particles. J. Geophys. Res. 1995, 100, 18753–18770.
- Folkers, M.; Menel, T.; Wahner, A. Influence of an organic coating on the reactivity of aqueous aerosols probed by heterogeneous hydrolysis of N2O. Geophys. Res. Lett. 2003, 30, 1644, doi:10.1029/2003GL017168.
- Ziemann, P.; Atkinson, R. Kinetics, products and mechanisms of secondary aerosol formation. Chem. Soc. Rev. 2012, 41, 6582–6605.
- Middleton, P.; Brock, J. Simulation of aerosol kinetics. J. Colloid Interface Sci. 1976, 54, 249–264.
- Griffin, R.; Dabdub, D.; Seinfeld, J. Secondary organic aerosol 1. Atmospheric chemical mechanism for production of molecular constituents. J. Geophys. Res. Atmos. 2002, 107, 3–26, doi:10.1029/2001JD000541.
- Pun, B.; Griffin, R.; Seigneur, C.; Seinfeld, J. Secondary organic aerosol 2. Thermodynamic model for gas/particle partitioning of molecular constituents. J. Geophys. Res. Atmos. 2002, 107, 4333, doi.10.1029/2001JD000542.
- Chan, A.; Kroll, J.; Ng, N.; Seinfeld, J. Kinetic modeling of secondary organic aerosol formation: Effects of particle-and gas-phase reactions of semivolatile products. Atmos. Chem. Phys. 2007, 7, 4135–4147.
- Li, J.; Cleveland, M.; Ziemba, L.; Griffin, E.; Barsanti, K.; Pankow, J.; Ying, Q. Modeling Regional Secondary Organic Aerosol Using the Master Chemical Mechanism. Atmos. Environ. 2015, 102, 52–61.
- Jathar, S.; Cappa. C.; Wexler, A.; Seinfeld, J.; Kleeman, M. Multidimensional oxidation model to simulate secondary organic aerosol in a 3-D air quality model. Geosci. Model Dev. 2015, 8, 2553–256.
- US Environmental Protection Agency. Mechanisms in the CMAQv5.1 Model. Available online: www.airquality modeling.org/index.php/CMAQv5.1_Mechanisms (accessed on 5 June 2019).
- Vernecek, M.; Cai, C.; Kaduwela, A.; Avise, J.; Carter, W.; Kleeman, M. Analysis of SAPRC16 chemical mechanism for ambient simulations. Atmos. Environ. 2018, 192, 136–150.
- National Center for Atmospheric Research (NCAR). Generator of Explicit Chemistry and Kinetics of Organics in the Atmosphere (GECKO-A). Available online: www2.acom.ucar.edu/modeling/ecko (accessed on 6 June 2019).
- Lannuque, V.; Camredon, M.; Couvidat, F.; Hodzic, A.; Valorso, R.; Madronich, S.; Bessagnet, B.; Aumont, B. Exploration of the influence of environmental conditions on secondary organic aerosol formation and organic species properties using explicit simulations: Development of the VBS-GECKO parameterization. Chem. Phys. Discuss. 2018, 18, 13411–13428, doi:10.5194/acp-2018-233.
- Praske, E.; Otkjaar, R.; Croiunse, J.; Hethcox, J.C.; Stoltz, B.; Kjaergaard, H. Atmospheric autoxidation is increasingly important in urban and suburban North America. Proc. Natl. Acad. Sci. USA 2018, 115, 64–69.
- Kim, Y.; Coudivat, F.; Sartelet, K.; Seigneur, C. Comparison of Different Gas-Phase Mechanisms and Aerosol Modules for Simulating Particulate Matter Formation. J. Air Waste Manag. Assoc. 2011, 61, 1218–1226.
- Stanier, C.O.; Donahue, N.M.; Pandis, S.N. Parameterization of secondary organic aerosol mass fractions from smog chamber data. Atmos. Environ. 2008, 42, 2276–229232. Hidy, G.M. Worldwide aerosol chemistry: From hemispheric distributions to megacity sources. J. Air Waste Manag. Assoc. 2009, 59, 770–789.
- Jimenez, M.; Canagarata, N.; Donahue, N.; Prevot, A.; Zhang, Q.; Kroll, J.; DeCarlo, P.; Allan, J.; Coe, H.; Ng, N.; et al. Evolution of Organic Aerosols in the Atmosphere. Science 2009, 326, 1525–1529.
- Turpin, B.; Huntzicker, J. Identification of Secondary Organic Aerosol Episodes and Quantification of Primary and Secondary Organic Aerosol Concentrations during SCAQS. Atmos. Environ. 1995, 29, 327–354.
- Zhang, Q.; Alfarra, M.; Worsnop, D.; Allan, J.; Coe, H.; Canagarata, M.; Jiminez, J. Deconvolution and quantification of hydrocarbon-like and oxygenated organic aerosols based on aerosol mass spectrometry. Environ. Sci. Technol. 2005, 39, 4938–4952.
- Rinaldi, M.; Gilardoni, S.; Paglione, M.; Sandrini, S.; Fuzzi, S.; Massoli, P.; Bonasoni, P.; Cristofanelli, P.; Marinoni, A.; Poluzzi, V.; et al. Organic aerosol evolution and transport observed at Mt. Cimone (2165 m asl), Italy, during the PEGASOS campaign. Atmos. Chem. Phys. 2015, 15, 11327–11240.
- Carlton, A.; DeGouw, J.; Jiminez, J.; Ambrose, J.; Attwood, A.; Brown, S.; Baker, K.; Brock, C.; Cohen, R.; Edgerton, S.; et al. Synthesis of the Southeast Atmosphere Studies. Bull. Am. Meteorol. Soc. 2018, 98, 547–567.
- Mao, J.; Carlton, A.; Cohen, R.; Brune, W.; Brown, S.; Wolfe, G.; Jiminez, J.; Pye, H.; Lee, Ng, N.; Xu, L.; et al. Southeast Atmosphere Studies: Learning from model-observation syntheses. Atmos. Chem. Phys. 2018, 18, 2615–2651.
- Kourtchev, I.; Ruukanen, T.; Maenhaut, W.; Kulmala, M.; Claeys, M. Observations of 2-methyltetrols and related photooxidation products of isoprene in boreal forest aerosols from Hyytiala, Finland. Atmos. Chem. Phys. 2005, 5, 2761–2770.
- Froyd, K.; Murphy, S.; Murphy, D.; deGouw, J.; Eddingrass, N.; Wennberg, P. Contribution of isoprene-derived organosulfates to free tropospheric aerosol mass. Proc. Natl. Acad. Sci. USA 2010, 107, 21360–21365.
- Zhang, H.; Worton, D.R.; Lewandowski, M.; Ortega, J.; Rubitschun, C.L.; Park, J.-H.; Kristensen, K.; Campuzano-Jost, P.; Day, D.A.; Jiménez, J.L.; et al. Organosulfates as Tracers for Secondary Organic Aerosol (SOA) Formation from 2-Methyl-3-Buten-2-ol (MBO) in the Atmosphere. Environ. Sci. Technol. 2012, 46, 9437–9446.
- Muller, L.; Reinnig, M.-C.; Naumann, K.; Saathoff, H.; Menetel, T.; Donahue, N.; Hoffmann, Y. Formation of 3-methyl-1,2,3-butanetricarboxylic acid via gas phase oxidation of pinonic acid-a mass spectrometric study of SOA aging. Atmos. Chem. Phys. 2012, 12, 1483–1496.
- M.; Cass, G.; Schauer, J.; Edgerton, E. Source apportionment of PM2.5 in the southeastern United States using solvent extractable organic compounds. Environ. Sci. Technol. 2001, 36, 2361–2371.
- Eschenroeder, A.; Deley, G.; Wahl, R. Field Program Designs for Verifying Photochemical Models; Report EPA R4-73-012c; Environmental Protection Agency: Research Triangle Park, NC, USA, 1973.
- Fiegley, C.; Jeffries, H.; Kamens, R. An Experimental Simulation of Los Angeles Reactive Pollutant Program (LARPP) Operation 33—Part 1. Experimental Simulation in an Outdoor Smog Chamber. Atmos. Environ. 1979, 16, 1989–1996.
- Fiegley, C.; Jeffries, H. Analysis of Processes Affecting Oxidant and Precursors in the Los Angeles Reactive Pollutant Program (LARPP) Operation 33. Atmos. Environ. 1979, 13, 1369–1384247. Hidy, G.M. An Historical Experiment: Los Angeles Smog Evolution Observed by Blimp. J. Air Waste Manag. Assoc. 2018, 68, 634–655, doi:10.1080/10962247.2018.1433251.
- Edinger, J. Vertical Distribution of Photochemical Smog in Los Angeles Basin. Environ. Sci. Technol. 1973, 7, 247–252.
- S. Environmental Protection Agency (EPA). Community Multiscale Air Quality Modeling System (CMAQ). Available online: www.epa.gov/cmaq (accessed on 12 April 2019).
- Wang, M.; Penner, J.; Liu, X. Coupled IMPACT aerosol and NCAR CAM3 model: Evaluation of predicted aerosol number and size distribution. J. Geophys. Res. Atmos. 2009, 114, doi:10.1029/2008jD010459.
- Kaiser, J.C.; Hendricks, J.; Righi, M.; Jöckel, P.; Tost, H.; Kandler, K.; Weinzierl, B.; Sauer, D.; Heimerl, K.; Schwarz, J.P.; et al. Global aerosol modeling with MADE-3 (v3.0) in EMAC (based on v2.53): Model description and evaluation. Geosci. Model Dev. 2019, 12, 541–579.
- Long, M.; Yantosca, J.; Nielsen, J.; Keller, C.; da Silva, M.; Pawson, S.; Jacob, D. Development of a Grid-independent GEOS-Chem Chemical Transport Model as an Atmospheric Chemistry Module or Earth System Models. Geosci. Model Dev. 2014, 7, 7505–7524.
- Solomon, P.; Cowling, E.; Hidy, G.; Furiness, C. Comparison of scientific findings from major ozone field studies and aerosol precursors. Atmos. Environ. 2000, 34, 1885–1920.
- Hidy, G.M. Atmospheric Sulfur and Nitrogen Oxides; Academic Press: San Diego, CA, USA, 1994.
- Hansen, D.A.; Edgerton, E.S.; Hartsell, B.; Jansen, J.; Hidy, G.; Kandaswamy, K.; Blanchard, C. The Southeastern Aerosol Research and Characterization study (SEARCH): 1. Overview. J. Air Waste Manag. Assoc. 2003, 53, 1460–1471.
- Solomon, P.; Sioutas, C. Continuous and semicontinuous monitoring techniques for particulate matter mass and chemical components: A synthesis of findings from EPA’s particulate matter supersites program and related studies. J. Air Waste Manag. Assoc. 2008, 58, 164–195.
- Woody, M.C.; Baker, K.R.; Hayes, P.L.; Jimenez, J.L.; Koo, B.; Pye, H.O. Understanding sources of organic aerosol during CalNex-2010 using the CMAQ-VBS. Atmos. Chem. Phys. 2016, 16, 4081–4100.
- Baker, K.; Kleindienst, T.; Offenberg, J.; Beaver, M.; Kelly, J.; Carlton, A. Evaluation of CMAQ Estimated Gas and Aerosol Carbon Using STN, Improve and CalNex Field Measurements. In Proceedings of the 13th Annual CMAS Conference, Research Triangle Park, NC, USA, 27–29 October 2014.
- Mebust, M.; Eder, B.; Binkowski, F.; Roselle, S. Models-3 Community Multiscale Air Quality (CMAQ) model aerosol component 2. Model Evaluation. J. Geophys. Res. 2003, 108, doi:10/1029/2001JD001410.
- Ciarelli, C.; Aksoyoglu, S.; Crippa, M.; Jiminez, J.; Nemitz, E. Evaluation of European air quality modelled by CAMx including the volatility basis set scheme. Atmos. Chem. Phys. 2016, 16, 10313–10332.
- Mircea, M.; Bessagnet, B.; D’Isidoro, M.; Pirovano, G.; Aksoyoglu, S.; Ciarelli, G.; Tsyro, S.; Manders, A.; Bieser, J.; Stern, R.; et al. EURODELTA III exercise: An evaluation of air quality models’ capacity to reproduce the carbonaceous aerosol. Atmos. Environ. X 2019, 2, 100018.
- New York Deptartment of Environmental Conservation. CMAQ Model Performance Assessment 8-Hr OTC Ozone Modeling; Report TSD-1e; Bureau of Air Quality and Research Division: Albany, NY, USA, 2006.
- Appel, K.; Chemel, C.; Roselle, S.; Frabcis, X.; Hu, T.-M.; Sokhl, R.; Rao, S.; Galmarini, S. Examination of Community Multiscale Air Quality (CMAQ) model performance over the North American and European domains. Atmos. Environ. 2012, 53, 142–155.
- Volkamer, R.; Jiminez, J.; San Martini, F.; Dzepina, K.; Salcedo, D.; Molina, L.; Worsnop, S.; Molina, M. Secondary organic aerosol formation from anthropogenic in pollution: Rapid and higher than expected. Geophys. Lett. 2006, 33, L17811.
- Nguyen, T.B.; Crounse, J.D.; Schwantes, R.H.; Teng, A.; Bates, K.H.; Zhang, X.; Clair, J.M.S.; Brune, W.H.; Tyndall, G.S.; Keutsch, F.N.; et al. Overview of the Focused Isoprene eXperiment at the California Institute of Technology (FIXCIT): Mechanistic chamber studies on the oxidation of biogenic compounds. Atmos. Chem. Phys. 2014, 14, 13531–13549.