1.1. DNA Damage and DDR
DNA carries genetic instructions for the development, functioning, growth, and reproduction of cells. DNA is inherently unstable due to both spontaneous chemical instability and modifications caused by either exogenous or endogenous agents causing DNA damage [
1,
2]. It has been estimated that each individual cell is subjected to up to one million DNA changes per day [
3,
4,
5,
6]. DNA damage is well known to affect both DNA replication, transcription, and a broad spectrum of signaling pathways including the nucleus to the mitochondria signaling pathway [
1,
7]. In this review, we update the progress of mechanistic studies on the key DNA repair pathways, including base excision repair (BER), nucleotide excision repair (NER), and double-strand break repair (DSBR). Furthermore, we focus on the role of DNA damage in ageing-related neurodegenerative diseases, with particular attention to its role in both rare premature ageing diseases, and the age-predisposed condition Alzheimer’s disease (AD).
Unlike other macromolecules of the cell, DNA cannot be replaced, but must be repaired to remain intact and functional. To avert deleterious consequences of DNA damage, cells have evolved several mechanisms, collectively termed the DNA damage response (DDR), to detect DNA damage, signal its presence and promote its repair. A variety of DDR pathways have been identified in organisms ranging from bacteria to humans, and are essential to life [
8]. Three main repair pathways in mammalian neurons, including base excision repair (BER), nucleotide excision repair (NER), and double strand break repair (DSBR), are discussed below.
1.2. Major DNA Repair Pathways and Their Roles in Neurons and Microglia
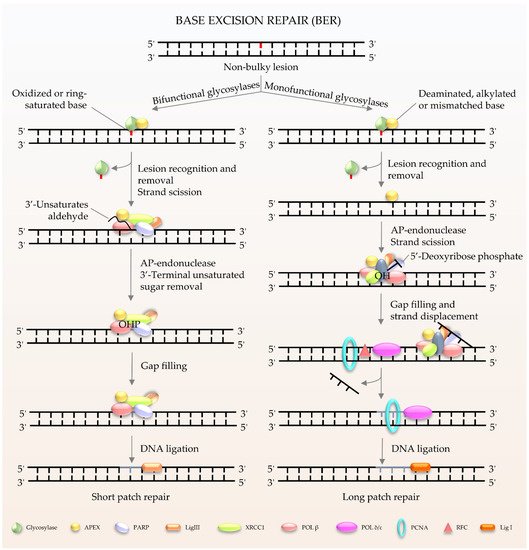
BER (
Figure 1) is the major pathway involved in the repair of oxidative lesions and is responsible for the repair of non-bulky DNA oxidation, deamination, and alkylation [
9,
10,
11]. The first step in the BER pathway uses a lesion-specific DNA glycosylase to recognize and eliminate damaged base pairs, which initiates the pathway [
12]. Either a bifunctional or monofunctional DNA glycosylase catalyzes the cleavage of the
N-glycosidic bond by flipping the damaged base out of the double helix to release a free base and create an abasic site (AP site). The repair is further processed by AP endonuclease 1 (APE1) to cleave the DNA backbone 5′ to the AP site, hereby producing 3′-hydroxyl and 5′-2-deoxyribose-5′-phosphate (5′-dRP). The gap is then filled by DNA polymerases using 3′-hydroxyl through template-directed synthesis via either short-patch or long-patch repair, depending on the number of inserted nucleotides. In the case of short-patch BER, DNA polymerase β (Polβ) and the inherent dRP-lyase activity of Polβ detaches the 5′-dRP to replace a single nucleotide. In general, long-patch repair requires the assistance of Polδ/ε and flap endonuclease 1 (FEN1) to substitute the displaced 5′-end structures with 2–13 nucleotides [
10]. The importance of BER has been exemplified by the lethality of
Ape1−/− or
Polβ−/− mouse models. Deletion of mouse APE1 (also known as Apex1) leads to embryonic lethality, and deficiency in cells can promote cellular senescence and premature ageing features [
13]. It has been demonstrated that Polβ defects can hamper amyloid β (Aβ)-induced neurogenesis in mice [
14]. In particular, knockdown of Polβ inhibited the 42 amino acids form of Aβ peptide (Aβ
1–42)-promoted differentiation of nestin
+ progenitor cells into nestin
+/Distal-less homeobox 2 (Dlx-2
+) neuroblasts [
14]. Moreover, pharmacological blockage of Polβ prevented Aβ
1–42-induced differentiation of progenitors into MAP-2
+ neurons [
14]. It is worth noting that many proteins involved in BER not only exist in the nucleus, but also in the mitochondria [
8]. Mitochondrial DNA (mtDNA) is more susceptible to the adverse effects of reactive oxygen species (ROS) produced during oxidative phosphorylation than nuclear DNA [
15]. Thus, BER is considered to be the main repair pathway for DNA damage in mitochondria [
16,
17].
Figure 1. Schematic of BER in mammalian cells. BER is the key pathway to remove and repair damaged bases. It starts with a DNA glycosylase to recognize and eliminate the damaged base, creating an abasic site. The gap is finally filled by DNA polymerases. The short patch is repaired with a single nucleotide, and the long patch is synthesized with 2–13 nucleotides.
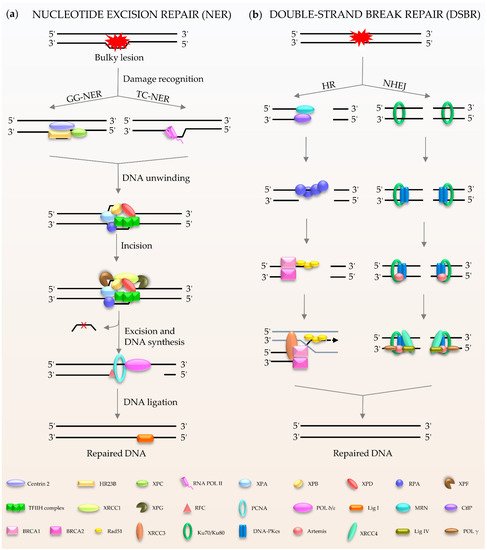
NER (
Figure 2a) is a central DNA repair pathway and a highly dynamic process responsible for removing bulky lesions from the genome [
18,
19,
20]. In humans, NER is composed of two branches, global genome NER (GG-NER) [
21] and transcription-coupled NER (TC-NER) [
22,
23], which are distinguished by their different recognition methods. XPC, as the main damage recognition factor, scans the double helix to identify the lesion, and forms a complex with centrins, HR23B, etc., to induce GC-NER activity [
24]. In the case of TC-NER, it is primed by transcribing RNA polymerase II [
25]. After recognition, the two factors share the same pathway for repairing the damage. The repair factor XPA and the large multi-subunit transcription factor IIH (TFIIH) act as translocase and helicase, respectively, to unwind the DNA [
19,
26]. The DNA lesion is then excised through double DNA nicks with two endonucleases, XPF and XPG. Finally, the gap is filled by Polδ/ε and ligase I [
21,
22].
Figure 2. Schematic of NER and DSBR in mammalian cells. (a) NER is composed of GG-NER and TC-NER. XPC scans the double helix to identify the lesion, and forms a complex with centrins, HR23B among others, to induce NER activity. Subsequently XPA and TFIIH act as translocase and helicase to unwind the DNA. The DNA lesion is excised by XPF and XPG. Finally, the gap is filled by DNA polymerases and DNA ligase I. (b) DSBR consists HR and NHEJ. HR is initiated by MRN. Rad51 participates in the search for homologous copies, and its homologues are involved in DNA strand invasion and subsequent homologous recombination to repair. NHEJ is activated by Ku80/Ku70. The damaged ends are trimmed by artemis; after that, the gap is filled by the action of DNA polymerase. The final repair is mediated by DNA ligase IV and cofactor XRCC4.
Double-stranded breaks (DSBs) are the most harmful type of DNA damage in terms of genomic integrity [
27]. Mammalian cells use two main DSBR pathways: homologous recombination (HR) and non-homologous DNA end joining (NHEJ) (
Figure 2b) [
28,
29,
30,
31,
32]. HR is the main method for DSBR utilized during embryogenesis and embryonic development [
33]. The damaged ends of DNA are recognized by the Mre11-Rad50-Nbs1 (MRN) complex [
34]. This complex performs extensive DNA processing, which together with CtIP generates 3′ single-stranded DNA (ssDNA). Rad51 participates in the search for homologous copies, and its homologues are involved in DNA strand invasion and subsequent HR to achieve highly accurate damage repair [
35]. The cyclic heterodimer Ku70/Ku80 triggers NHEJ and then recruits DNA-PKcs [
36,
37,
38,
39]. Afterwards, nucleases (such as Artemis) deal with the damaged ends, and the gap is filled by DNA polymerases, such as Pol μ/λ/γ [
40,
41]. The final step of the repair pathway is mediated by DNA ligase IV and X-ray repair cross-complementing protein (XRCC4) [
39,
41]. It is well known that the process of DDR requires ATP, especially for DNA ligation. In humans, in response to DNA damage, cells mobilize more than ten thousand ATP molecules to repair just one DSB [
42]. Interestingly, DNA ligase IV, the key enzyme of DSBR, uses NAD
+ as a substrate for double-strand connection to mediate the final repair [
43].
Neuronal homeostasis is a prerequisite for the development and function of the nervous system, and requires high fidelity and stable inheritance [
44]. In normal cellular activities or DNA replication, the high precision and integrity of the genome must be maintained after DNA damage. Multiple DDR pathways in cells drive the biological functions ensuring this procedure. During the early stages of neuronal development, that is, neural progenitor proliferation, the nervous system has entire DDR pathways. In other words, it can repair double-strand breaks through two pathways of HR and NHEJ before neuronal maturation (
Figure 3) [
11,
44]. In contrast, during neuronal maturation, NHEJ becomes the only way to repair double-strand break damage [
45]. Defects in these DDRs can cause neurological disorders. Studies have shown that defects in NHEJ, NER, or BER increase risks to neurodegenerative disorders or neurodevelopmental defects [
46,
47,
48,
49,
50,
51].
Figure 3. Schematic of the availability of different DNA repair pathways during different stages of neuronal development.
Microglia are glial cells widely distributed in the brain and spinal cord. They are the main form of active immune defense in the central nervous system, and are key cells involved in the neuroprotective functions that maintain normal brain function; however, microglia can be hostile to neurons in disease conditions [
52,
53]. Studies have shown that DDR-related proteins have an impact on the activity, function, and survival status of microglia. In DDR, especially in DSBR, the deficiency of one crucial protein, ataxia-telangiectasia mutated (ATM) [
54], results in abnormally active microglia, and stimulates excessive production of pro-inflammatory factors, which result in neurotoxicity [
55]. More specifically, ATM dysfunction causes damage to DNA repair, and leads to the further accumulation of impaired cytoplasmic DNA. In microglia, cytoplasmic DNA can subsequently activate an antiviral defense system via the DNA sensor stimulator of interferon genes (STING). Cytoplasmic DNA can also trigger absent in melanoma 2 (AIM2) inflammasomes and, in parallel, induce elevated levels of cytokine precursors, such as pro-IL-1, through proteolytic processing. These processes create an extreme environment of neurotoxic inflammation [
55]. In addition, the DNA excision repair protein, ERCC1, is very important for NER and DSBR [
56]. Loss of ERCC1 results in microglial death and a compensatory increase in proliferation [
57]. Of note, it is speculated that, in a mouse model of
Cx3cr1-Ercc1ko/loxP and
Cx3cr1-Ercc1wt/loxP,
Ercc1-deficient microglia might have a link with ageing-related phenotypes [
57].