绝大多数胰腺导管腺癌患者的肿瘤中含有 KRAS 突变。在功能上,突变的KRAS不仅致力于肿瘤细胞的增殖,存活和侵袭性,而且还导致该癌症的免疫抑制。
- KRAS gene
- pancreatic ductal adenocarcinoma
- cancer immunity
- immune checkpoint blockade
一、简介
在人类中,胰腺导管腺癌(PDAC)患者通常预后较差。肿瘤中现有的免疫环境会影响ICB治疗的有效性[ 6 ]。在 PDAC 中,肿瘤环境通常具有免疫抑制作用 [ 7 ]。最近,人们已经认识到驱动癌基因在癌症免疫状态中发挥着令人信服的作用 [ 8 ]。在 PDAC 中,Kirsten 大鼠肉瘤病毒癌基因同源物 ( KRAS ) 基因发生广泛突变 [ 9 ]。PDAC 中的KRAS突变包括由密码子 12 或密码子 13 中的错义突变引起的突变,导致原始甘氨酸 (G) 被其他氨基酸取代,从而导致持续激活在这种情况下的KRAS蛋白 [ 9 ]。的KRAS突变作为驾驶员原因PDAC发生,发展与其它基因,如肿瘤蛋白P53基因(的伴随灭活TP53),细胞周期蛋白依赖性激酶抑制剂2A基因(CDKN2A)和SMAD家族成员4基因(SMAD4) [ 10 , 11 ](图 1)。在这个过程中,KRAS突变也会导致下游通路的激活,这些通路可以提高癌细胞的存活、增殖、免疫逃避和耐药性[ 7 , 9 ]。关于 PDAC 中的免疫抑制,KRAS突变利用多种途径来实现这一目标,例如激活 yes 相关蛋白 (YAP)-tafazzin (TAZ) 通路及其下游 Janus 激酶信号转导和转录激活因子 3 (JAK-STAT3) 信号传导 [ 12 ],诱导细胞通过重新编程的葡萄糖代谢[自噬相关的主要组织相容性复合体I(MHC-I)降解13,14 ],并与其他的遗传改变协同(例如,TP53失活)[ 15 ](图1)。因此,PDAC 肿瘤可以被具有促癌功能的骨髓细胞浸润,例如中性粒细胞、骨髓源性抑制细胞 (MDSCs) 和 M2 样巨噬细胞 [ 7]]。
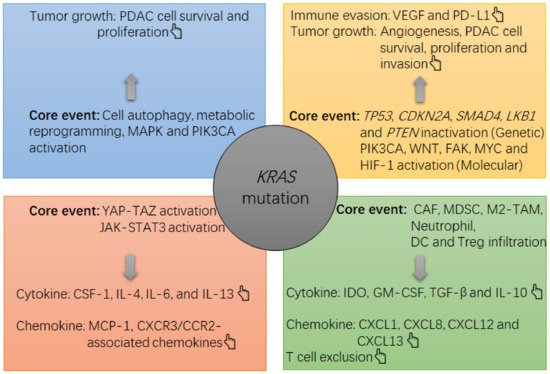
图 1. PDAC 肿瘤中KRAS突变诱导生长和免疫抑制的注释图。该KRAS突变主要通过以下途径在 PDAC 肿瘤中引起抑制环境,例如激活丝裂原活化蛋白激酶 (MAPK) 和磷脂酰肌醇 3-激酶 (PI3K)-Akt,激活 YAP-TAZ 和 JAK-STAT3,以及诱导PDAC细胞中的细胞自噬和代谢重编程。在这种情况下,PDAC细胞的存活和增殖会加速,肿瘤细胞的过度生长会导致肿瘤内缺氧,进而激活缺氧诱导因子1(HIF-1)α,上调编码基因的表达。血管内皮生长因子 (VEGF) 通过 PDAC 细胞。VEGF 是一种有效的细胞因子,可诱导血管生成和免疫逃避(例如,程序性死亡配体 1 (PD-L1) 上调和杀伤肿瘤性 T 细胞耗竭)。同时,PDAC细胞可以增加抑制性细胞因子和趋化因子的产生,例如白介素4(IL-4),IL-6,IL-13,巨噬细胞集落刺激因子1(CSF-1)和单核细胞趋化蛋白1(MCP-1) ),然后招募并增加免疫浸润物的存活和抑制功能,包括癌症相关成纤维细胞 (CAF)、MDSC、M2 样肿瘤相关巨噬细胞 (TAM)、产生吲哚胺 2, 3-双加氧酶 (IDO) 的树突细胞(DC) 和调节性 T 细胞 (Treg 细胞)。在这种情况下,抑制性细胞的过载将增加抑制性细胞因子和趋化因子的局部水平,例如转化生长因子-β (TGF-β)、IDO、IL-10、粒细胞-巨噬细胞集落刺激因子 (GM-CSF) , 趋化因子 CXC 基序配体 1 (CXCL1)、CXCL8、CXCL12 和 CXCL13,从而加强肿瘤中的免疫抑制(例如,杀瘤性 T 细胞排除)。与KRAS突变,基因和分子水平的其他改变,例如肝激酶 B1 基因 ( LKB1 ) 失活、TP53失活、磷酸酶和张力蛋白同源基因 ( PTEN ) 失活、粘着斑激酶 (FAK) 激活、磷脂酰肌醇 4, 5-二磷酸3-激酶催化亚基 α (PIK3CA) 激活或 Wingless/整合 (WNT) 激活也有助于肿瘤生长(例如 PDAC 细胞存活、增殖和侵袭)和免疫逃避(PD-L1 上调)。
2. KRAS突变在 PDAC 中的致癌作用
人PDAC只具有KRAS突变而不是神经母细胞瘤RAS病毒癌基因同系物基因(NRAS)或哈维大鼠肉瘤病毒原癌基因同源基因(HRAS)突变[ 9 ]。总体而言,97.7% 的 PDAC 病例被检测到具有KRAS突变 [1][2][3][4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82][83][84][85][86][87][88][89][90][91][92][93][94][95]。G12D、G12V和G12R是PDAC中三种最常见的KRAS突变错义形式,其中以G12D错义突变最为常见[ 9 ](表1)。生理上,正常的KRAS蛋白具有 GTPase 活性,但这些错义变体会产生与 GTP 稳定结合的 KRAS 蛋白,从而组成性激活 MAPK 和 PI3K-Akt 通路,这两条经典通路负责维持细胞存活和增殖 [ 35 ](图 1)。
| 癌症 | 数模转换器 | CRAC | LUAC | |
|---|---|---|---|---|
| 字符 [参考] | ||||
| KRAS突变的流行率 | 97.7% [ 9 ] | 44.7% [ 9 ] | 30.9% [ 9 ] | |
| KRAS 中最热门的错义突变 | G12D [ 9 ] | G12D [ 9 ] | G12C [ 9 ] | |
| Sensitive to glucose restriction vs. KRASwt | Yes [19] | Yes [20] | No [21] | |
| Common alteration with KRAS | TP53 inactivation [10] | TP53 and APC inactivation [22] | TP53 or LKB1 inactivation [23] | |
| General milieu of KRAS-mutant tumors | Immune-cold [7] | Immune-cold [24] | KRAS-only: immune-cold or hot [23] TP53 inactivation: immune-hot [23] LKB1 inactivation: immune-cold [23] |
|
| Number/function of tumoricidal T cells in KRAS-mutant tumors | Decrease/Decrease [7] | Decrease/Decrease [24] | KRAS-only: slight increase/decrease [23] TP53 inactivation: significant increase/decrease [23] LKB1 inactivation: significant decrease/decrease [23] |
|
| Major type of immune infiltrates in KRAS-mutant tumors | Myeloid suppressive cell [7] | Myeloid suppressive cell [24] | KRAS-only: T cell, macrophage, neutrophil [23] TP53 inactivation: CD8+ T cell, CD45RO+ T cell [23] LKB1 inactivation: myeloid suppressive cell [23] |
|
| Common presentation of the ICB therapy biomarker if KRAS mutation | pMMR/MSS [25] | pMMR/MSS [26] | KRAS-only: PD-L1 expression ↑ [23] TP53 inactivation: PD-L1 expression ↑↑ [23] LKB1 inactivation: PD-L1 expression ↓↓ [23] |
|
| Biomarker associated with the effectiveness of ICB therapy | dMMR/MSI-H [4] | dMMR/MSI-H [27] | PD-L1 [23] | |
| Prevalence of dMMR/MSI-H in all cases | 1~2% [25] | 14% [26] | NM | |
| Prevalence of positive expression of PD-L1 by tumor cells | NM | NM | Among KRAS-only tumors: 37.5% [23] Among TP53 inactivation tumors: 68.8% [23] Among LKB1 inactivation tumors: 10% [23] |
|
| General response to monotherapy using ICB drugs | Poor [5] | Poor [28] | KRAS-only tumor: Fair [23] TP53 inactivation tumor: Excellent [23] LKB1 inactivation tumor: Poor [23] |
|
| Core molecular events associated with KRAS mutation-induced immunosuppression | 1. YAP-TAZ activation [12]; 2. JAK-STAT3 activation [12]; 3. Metabolic reprogramming of glucose and cell autophagy [13,14]; 4. In concert with other events, TP53 inactivation [15], LKB1 mutation [29,30], PTEN loss [29,30], WNT/β-catenin activation [29,30], FAK activation [29,30], PIK3CA activation [29,30] and MYC activation [29,30]; |
1. In concert with APC and TP53 inactivation: TGF-β1 upregulation and EMT [31]; 2. TGF-β-induced immune suppression [32]; 3. IRF2 inactivation [24,33]; 4. Metabolic dysregulation in glucose, glutamine, fatty acid and lipid [26,34]; 5. MAPK and HIF-1-related cascade activation [34]; |
1. ERK activation-induced PD-L1 upregulation [23] 2. Metabolic reprogramming of glucose [21] 3. In concert with LKB1 inactivation: strengthening metabolic reprogramming of glucose and JAK-STAT3 activation [23] |
|
3. The KRAS Mutation and Immune Environment in PDAC
In addition to impacting cell survival, proliferation and nutrient metabolism during pancreatic carcinogenesis, KRAS mutations also function in controlling the cancer immune environment. In addition, in mice bearing pancreatitis-induced ADM, KLF5 deficiency was revealed to suppress STAT3 activation [36]. In another mechanism, KRASG12D mutation-induced upregulation of YAP and TAZ was revealed to potently activate the downstream JAK-STAT3 pathway during pancreatic carcinogenesis in mice [12] (Figure 1). In fact, mutant KRAS can cooperate with extracellular stimuli, such as inflammation, the gut microbiota and gastrointestinal peptides, to persistently activate downstream YAP-TAZ signaling, which undermines immune surveillance against PDAC cells in addition to improving their proliferation, invasion, survival and metabolism [42]. In PDAC, a high expression of YAP was revealed to correlate with a poor histological grade of tumor cells [43], a high risk of metastasis and a poor prognosis of patients [44].
Mechanistically, KRAS mutation-induced activation of YAP enables PDAC cells to release IL-4, IL-6, IL-13, MCP-1 and CSF-1, which promote the recruitment of tumor-associated macrophages (TAMs) into tumors and induce them to proliferate and polarize into an M2-like phenotype [45] (Figure 1). In addition, the prevalence of TP53 inactivation is only second to the prevalence of KRAS mutation in PDAC [10], meaning that a large portion of patients concomitantly harbor KRAS mutation and TP53 inactivation [10].
In concert with the KRAS mutation, alterations in environmental, genetic and molecular levels, such as hypoxia, LKB1 mutation, PTEN loss, PIK3CA activation, WNT/β-catenin activation, FAK activation and MYC proto-oncogene (MYC) activation also contribute to immune suppression in PDAC tumors [29,30] (Figure 1).
4. Current Status of Immune Checkpoint Blockade Therapy for PDAC
Since the tumoral milieu of PDAC is immunosuppressive, ICB therapy is anticipated to have low effectiveness in this cancer. In fact, several lines of clinical data have confirmed this speculation, and the effectiveness of monotherapy by using ICB drugs in patients with metastatic PDAC remains disappointing [5]. ICB drugs are not effective in significantly shrinking the size of PDAC tumors when used as a second- or later-line therapy regardless of whether they are used alone or in combination with radiotherapy or chemotherapy (Table 2).
| Author [Ref.] | Year | Phase | Patient No. | ICB Drug | Other Treatment | ORR |
|---|---|---|---|---|---|---|
| • First-line therapy | ||||||
| Aglietta M, et al. [62] | 2014 | I | 34 | Tremelimumab | Gemcitabine | 10.5% |
| Wainberg ZA, et al. [63] | 2019 | I | 50 | Nivolumab | Gemcitabine + Nab- paclitaxel | 18% |
| Wainberg ZA, et al. [64] | 2017 | I | 17 | Nivolumab | Gemcitabine + Nab- paclitaxel | 50% |
| Renouf, et al. [65] | 2018 | II | 11 | Durvalumab + Tremelimumab | Gemcitabine + Nab-paclitaxel | 73% |
| Borazanci, et al. [66] | 2018 | II | 11 | Nivolumab | Gemcitabine + Nab-paclitaxel + Cisplatin + Paricalcitol | 80% |
| • Second- or later-line therapy | ||||||
| Luke JJ, et al. [57] | 2018 | I | 3 | Pembrolizumab | SBRT: 30–50 Gy for 2–4 metastatic lesions | NR |
| O’Reilly EM, et al. [56] | 2019 | II | Arm A: 32 Arm B: 32 |
Durvalumab Durvalumab + Tremelimumab |
No | 0% 3.1% |
| Xie C, et al. [58] | 2020 | I | Arm A1: 14 Arm A2: 10 Arm B1: 19 Arm B2: 16 |
Durvalumab Durvalumab Durvalumab + Tremelimumab Durvalumab + Tremelimumab |
SBRT: 8 Gy/1 fraction SBRT: 25 Gy/5 fractions SBRT: 8 Gy/1 fraction SBRT: 25 Gy/5 fractions |
5.1% A |
| Weiss GJ, et al. [60] | 2017 | I | 11 | Pembrolizumab | Gemcitabine (Gem)-based chemotherapy | 18.2% |
| Kamath SD, et al. [61] | 2020 | I | 21 B | Arm A: Ipilimumab 3 mg/kg Arm B: Ipilimumab 3 mg/kg Arm C: Ipilimumab 6 mg/kg |
Gem 750 mg/m2 Gem 1g/m2 Gem 1g/m2 |
14% C |
5. Conclusions
PDAC 中的肿瘤环境具有很强的免疫抑制作用,这使得使用 ICB 药物的单一疗法几乎完全无效。关于 PDAC 免疫抑制的发展,涉及多种因素。在本文中,KRAS突变已被证明是该过程的核心,因为KRAS突变可以激活 YAP-TAZ 和 JAK-STAT3 以引发免疫抑制反应,然后可以通过与TP53失活和其他遗传或分子的协调来加强这种初始信号。改动。总体而言,KRAS突变通常与 PDAC 中的肿瘤免疫抑制相关。尽管如此,在 CRAC 和 LUAC 中,KRAS突变可以以不同的方式决定癌症免疫环境。在这些癌症中,尽管KRAS突变具有共性,但免疫环境各不相同。这个概念可以用KRAS 突变体 LUAC来举例说明,它对 ICB 治疗表现出不同的反应,这取决于与KRAS突变同时发生的基因改变的类型。
This entry is adapted from the peer-reviewed paper 10.3390/cancers13102429
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [CrossRef]
- Vincent, A.; Herman, J.; Schulik, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [CrossRef]
- Sohal, D.P.; Kennedy, E.B.; Cinar, P.; Conroy, T.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Lau, M.W.; Johnson, T.; Krishnamurthi, S.; et al. Metastatic Pancreatic Cancer: ASCO Guideline Update. J. Clin. Oncol. 2020, 27, 3217–3230. [CrossRef][PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; di Giacomo, A.M.; de Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J Clin. Oncol. 2020, 38, 1–10. [CrossRef] [PubMed]
- Henriksen, A.; Dyhl-Polk, A.; Chen, I.; Nielsen, D. Checkpoint inhibitors in pancreatic cancer. Cancer Treat. Rev. 2019, 78, 17–30. [CrossRef] [PubMed]
- Ribas, A.;Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [CrossRef] [PubMed]
- Liu, X.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Meng, Q.; Hua, J.; Yu, X.; Shi, S. The reciprocal regulation between host tissue and immune cells in pancreatic ductal adenocarcinoma: New insights and therapeutic implications. Mol. Cancer. 2019, 18, 184. [CrossRef] [PubMed]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS:Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [CrossRef]
- [10] Bailey P., Chang D. K. Nones K., Johns A. L., Patch A. M. Gingras M. C., et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016, 531: 47-52.
- [11] Kamisawa T., Wood L. D. Itoi T., Takaori K. Pancreatic cancer. Lancet. 2016, 388:73-85.
- [12] Gruber R., Panayiotou R., Nye E., Spencer-Dene B., Stamp G., Behrens A. YAP1 and TAZ Control Pancreatic Cancer Initiation in Mice by Direct Up-regulation of JAK-STAT3 Signaling. Gastroenterology. 2016, 151:526-39.
- [13] Humpton T. J., Alagesan B., DeNicola G. M., Lu D., Yordanov G. N. Leonhardt C. S., et al. Oncogenic KRAS Induces NIX-Mediated Mitophagy to Promote Pancreatic Cancer. Cancer Discov. 2019, 9:1268-1287.
- [14] Yamamoto K., Venida A., Yano J., Biancur D. E., Kakiuchi M., Gupta S., et al. Autophagy promotes immune evasion of pancre-atic cancer by degrading MHC-I. Nature. 2020, 581:100-105.
- [15] Blagih J., Zani F., Chakravarty P., Hennequart M., Pilley S., Hobor S., et al. Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep. 2020, 30:481-496.
- [16] Dong Z.Y., Zhong W. Z., Zhang X. C., Su J., Xie Z., Liu S. Y., et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin Cancer Res. 2017, 23:3021-3024.
- [17] Skoulidis F., Goldberg M. E., Greenawalt D. M., Hellman M. D. Awad M. M. Gainor J. F. et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8:822-835.
- [18] Liang T., Tong W., Ma S., Chang P. Standard therapies: solutions for improving therapeutic effects of immune checkpoint in-hibitors on colorectal cancer. OncoImmunology. 2020, 9:1773205.
- [19] Waters A. M., Der C. J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb Perspect Med. 2018, 8: a0311435.
- [20] He P., Yang J. W., Yang V. W., Bialkowska. A. B. Krüppel-like Factor 5, Increased in Pancreatic Ductal Adenocarcinoma, Pro-motes Proliferation, Acinar-to-Ductal Metaplasia, Pancreatic Intraepithelial Neoplasia, and Tumor Growth in Mice. Gastroen-terology. 2018, 154: 1494-1508.
- [21] Zhang W., Nandakumar N., Shi Y., Manzano M., Smith A., Graham G., et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci Signal. 2014, 7:ra42.
- [22] DeNicola G. M., Karreth F. A., Humpton T. J., Gopinathan A., Wei C., Frese K., Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011, 475:106-9.
- [23] Chang C., Qiu J., O’Sullivan D., Buck M. D. Noguchi T., Curtis J. D., et al. Metabolic Competition in the Tumor Microenviron-ment Is a Driver of Cancer Progression. Cell. 2015, 162:1229-41.
- [24] Kottakis F., Nicolay B. N., Roumane A., Karnik R., Gu H., Nagle J. M., et al. LKB loss links serine metabolism to DNA methyla-tion and tumorigenesis. Nature. 2016, 539: 390-395.
- [25] Jones S. A., Jenkins B. J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat Rev Immunol. 2018, 18:773-789.
- [26] Corcoran R. B., Contino G., Deshpande V., Tzatsos A., Conrad C., Benes C. H. et al. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011, 71:5020-9.
- [27] Eibl G., Rozengurt E. KRAS, YAP, and obesity in pancreatic cancer: A signaling network with multiple loops. Semin Cancer Biol. 2019, 54: 50-62.
- [28] Zhao X., Wang X., Fang L., Lan C., Zheng X., Wang Y., et al. A combinatorial strategy using YAP and pan-RAF inhibitors for treating KRAS-mutant pancreatic cancer. Cancer Lett. 2017, 402: 61-70.
- [29] Allende M., Zeron-Medina J., Hernandez J., Macarulla T., Balsells J., Merino X., et al. Overexpression of Yes Associated Protein 1, an Independent Prognostic Marker in Patients With Pancreatic Ductal Adenocarcinoma, Correlated With Liver Metastasis and Poor Prognosis. Pancreas. 2017, 46:913-920.
- [30] Yang W., Yang S., Zhang F., Cheng F., Wang X., Rao J. Influence of the Hippo-YAP signaling pathway on tumor associated macrophages (TAMs) and its implications on cancer immunosuppressive microenvironment. Ann Transl Med. 2020, 8:399.
- [31] Hingorani S. R., Wang L., Multani A. S., Combs C., Deramaudt T. B. Hruban R. H., et al. Trp53R172H and KRASG12D cooper-ate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005, 7:469-83.
- [32] Murakami S., Shabhazian D., Surana R., Zhang W., Chen H., Graham G. T., Yes-associated protein mediates immune repro-gramming in pancreatic ductal adenocarcinoma. Oncogene. 2017, 36:1232-1244.
- [33] Gan L. L., Hii L.W., Wong S. F., Leong C. O., Mai C. W. Molecular Mechanisms and Potential Therapeutic Reversal of Pancreatic Cancer-Induced Immune Evasion. Cancers (Basel). 2020, 12:1872.
- [34] Sivaram N., McLaughlin P. A., Han H. V., Petrenko O., Jiang Y. P., Ballou L. M., et al. Tumor-intrinsic PIK3CA represses tumor immunogenecity in a model of pancreatic cancer. J Clin Invest. 2019, 129:3264-3276.
- [35] Fukumura D., Kloepper J., Amoozgar Z., Duda D. G., Jain R. K. Enhancing cancer immunotherapy using antiangiogenics: op-portunities and challenges. Nat Rev Clin Oncol. 2018, 15:325-340.
- [36] Li S., Xu H. X., Wu C. T., Wang W. Q., Jin W., Gao H. L., et al. Angiogenesis in pancreatic cancer: current research status and clinical implications. Angiogenesis. 2019, 22:15-36.
- [37] Sodir N.M., Kortlever R. M., Barthet V. J. A., Campos T., Pellegrinet L., Kupczak S., et al. MYC Instructs and Maintains Pancre-atic Adenocarcinoma Phenotype. Cancer Discov. 2020, 10:588-607.
- [38] Lu C., Paschall A. V., Shi H., Savage N., Waller J.L., Sabbatini M. E., et al. The MLL1-H3K4me3 Axis-Mediated PD-L1 Expres-sion and Pancreatic Cancer Immune Evasion. J. Natl. Cancer Inst. 2017, 109: djw283.
- [39] Johnson B. A. 3rd., Yarchoan M., Lee V., Laheru D. A., Jaffee E. M. Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin Cancer Res. 2017, 23:1656-1669.
- [40] Joyce J. A., Fearon D. T. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015, 348: 74-80.
- [41] Feig C., Jones J. O., Kraman M., Wells R. J. B., Deonarine A., Chan D. S., Targeting CXCL12 from FAP-expressing carcino-ma-associated fibroblasts synergized with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013, 110: 20212-7.
- [42] Beatty G. L., Winograd R., Evans R. A., Long K. B., Luque S. L., Lee J. W., et al. Exclusion of T cells From Pancreatic Carcinomas in Mice Is Regulated by Ly6C(low) F4/80(+) Extracuumoral Macrophages. Gastroenterology. 2015, 149:201-10.
- [43] O’Reilly E.M., Oh D.Y., Dhani N., Renouf D.J. Lee M. A., Sun W., et al. Durvalumab With or Without Tremelimumab for Pa-tients With Metastatic Pancreatic Ductal Adenocarcinoma: A phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5: 1431-8.
- [44] Luke J.J., Lemons J.M., Karrison T. G., Pitroda S.P., Melotek J. M., Zha Y. Y., et al. Safety and Clinical Activity of Pembrolizumab and Multisite Stereotactic Body Radiotherapy in Patients With Advanced Solid Tumors. J Clin Oncol. 2018, 36:1611-1618.
- [45] Xie C., Duffy A. G., Brar G., Fioravanti S., Mabry-Hrones D., Walker M., et al. Immune Checkpoint Blockade in Combination with Stereotactic Body Radiotherapy in Patients with Metastatic Pancreatic Ductal Adenocarcinoma. Clin Cancer Res. 2020, 26:2318-2326.
- [46] Wang Q., Ju X., Wang J., Fan Y., Ren M., Zhang H. Immunogenic cell death in anticancer chemotherapy and its impact on clini-cal studies. Cancer Lett. 2018, 438: 17-23.
- [47] Weiss G. J., Waypa J., Blaydorn L., Coats J., McGahey K., Sangal A., et al. A phase Ib study of pembrolizumab plus chemother-apy in patients with advanced cancer (PmebroPlus). Br J Cancer. 2017, 117:33-40.
- [48] Kamath S. D., Kalyan A., Kircher S., Nimeiri H., Fought A.J., Benson 3rd Al., et al. Ipilimumab and Gemcitabine for Advanced Pancreatic Cancer: A phase Ib Study. Oncologist. 2020, 25: e808-e815.
- [49] Dawood S., Austin L., Cristofanilli M. Cancer stem cells: implications for cancer therapy. Oncology (Williston Park). 2014, 28:1101-7.
- [50] Riaz N., Havel J. J., Makarov V., Desrichard A., Urba W. J., Sims J. S., et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell. 2017, 171: 934-949.
- [51] Conroy T., Desseigne F., Ychou M., Bouché O., Guimbaud R., Bécouarn Y., et al. FOLFIRINOX versus gemcitabine for meta-static pancreatic cancer. N Engl J Med. 2011, 364:1817-25.
- [52] Von Hoff D. D., Ervin T., Arena F. P., Chiorean E. G., Infante J., Moore M., et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013, 369:1691-703.
- [53] Schumacher T.N., Scheper W., Kvistborg P. Cancer Neoantigens. Annu Rev Immunol. 2019, 37:173-200.
- [54] Aglietta M., Barone C., Sawyer M. B., Moore M. J., Miller Jr W. H., Bagalà C., et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann Oncol. 2014, 25:1750-5.
- [55] Wainberg Z. A., Hochster H.S., Kim E., George B., Kalyan A., Chiorean E.G., et al. Phase I study of nivolumab (nivo) + nab-paclitaxel (nab-P) + gemcitabine (Gem) in advanced pancreatic cancer (APC). J Clin Oncol. 2019, 37:298.
- [56] Wainberg Z. A., Hochster H. S., George B., Gutierrez M., Johns M. E., Chiorean E. G., et al. Phase I study of nivolumab (nivo) + nab-paclitaxel (nab-P) ± gemcitabine (Gem) in solid tumors: interim results from the pancreatic cancer (PC) cohorts [abstracts]. J Clin Oncol. 2017, 35: 412.
- [57] Renouf D.J., Dhani N.C., Kavan P., Jonker D.J., Wei AC-C., Hsu T., et al. The Canadian Cancer Trials Group PA.7 trial: results from the safety run in of a randomized phase II study of gemcitabine (GEM) and nab-paclitaxel (Nab-P) versus GEM, nab-P, durvalumab (D), and tremelimumab (T) as first-line therapy in metastatic pancreatic ductal adenocarcinoma (mPDAC). J Clin Oncol. 2018, 36: 349.
- [58] Borazanci E.H., Jameson G.S., Borad M.J., Ramanathan R.K., Korn R.L., Caldwell L., et al. A phase II pilot trial of nivolumab (N) + albumin bound paclitaxel (AP) + paricalcitol (P) + cisplatin (C) + gemcitabine (G) (NAPPCG) in patients with previously un-treated metastatic pancreatic ductal adenocarcinoma (PDAC). J Clin Oncol. 2018, 36:358.
- [59] Luchini C., Brosens L. A. A., Wood L. D. Chatterjee D., Shin J. II., Sciammarella C., et al. Comprehensive characterization of pancreatic ductal adenocarcinoma with microsatellite instability: histology, molecular pathology and clinical implications. Gut. 2020. Online ahead of print.
- [60] Allen E., Jabouille A., Rivera Lee B., Lodewijckx I., Missiaen R., Steri V., et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates immunity through HEV formation. Sci Transl Med. 2017, 9: eaak9679.
- [61] Mace T. A., Shakya R., Pitaarresi J. R., Swanson B., McQuinn C. W., Loftus S., et al. IL-6 and PD-L1 antibody blockade combina-tion therapy reduces tumour progression in murine models of pancreatic cancer. Gut. 2018, 67:320-332.
- [62] Zhang Q., Green M. D., Lang X., Lazarus J., Parsels J. D., Wei S., Inhibition of ATM Increases Interferon Signaling and Sensitizes Pancreatic Cancer to Immune Checkpoint Blockade Therapy. Cancer Res. 2019, 79:3940-3951.
- [63] Zhu Y., Knolhoff B.L., Meyer M.A., Nywening T.M., West B.L., Luo J., et al. CSF1/CSF1R blockade reprograms tu-mor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74:5057-69.
- [64] Stewart R. A., Pilié P.G., Yap T. A. Development of PARP and Immune-Checkpoint Inhibitor Combinations. Cancer Res. 2018, 78: 6717-6725.
- [65] Keenan B. P., Saenger Y., Kafrouni M. I., Leubner A., Lauer P., Maitra A., et al. A Listeria vaccine and depletion of T-regulatory cells activate immunity against early stage pancreatic intraepithelial neoplasms and prolong survival of mice. Gastroenterology. 2014, 146: 1784-94.
- [66] Jiang H., Hegde S., Knolhoff B. L., Zhu Y., Herndon J. M., Meyer M. A., et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016, 22:851-60.
- [67] Sanford D. E., Belt B. A., Panni R. Z., Mayer A., Deshpande A. D., Carpenter D., et al. Inflammatory monocyte mobilization de-creases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. 2013, 19:3404-15.
- [68] Blair A.B., Kleponis J., Thomas D.L. 2nd., Muth S. T., Murphy A. G., Kim V., et al. IDO1 inhibition potentiates vaccine-induced immunity against pancreatic adenocarcinoma. J Clin Invest. 2019, 129:1742-1755.
- [69] Burrack A. L., Spartz E. J., Raynor J. F., Wang I., Olson M., Stromnes I.M. Combination PD-1 and PD-L1 Blockade Promotes Durable Neoantigen-Specific T Cell-Mediated Immunity in Pancreatic Ductal Adenocarcinoma. Cell Rep. 2019, 28:2140-2155.
- [70] Christenson E. S., Jaffee E., Azad N. S. Current and emerging therapies for patients with advanced pancreatic ductal adenocar-cinoma: a bright future. Lancet Oncol. 2020, 21: e135-e145.
- [71] Nollmann FI., Ruess DA. Targeting Mutant KRAS in Pancreatic Cancer: Futile or Promising? Biomedicines. 2020,8(8):281.
- [72] Canon J., Rex K., Saiki A.Y., Mohr C., Cooke K., Bagal D., et al. The clinical KRAS(G12C) inhibitor AMG510 drives anti-tumour immunity. Nature. 2019, 575(7781):217-223.
- [73] Hallin J., Engstrom L.D., Hargis L., Calinisan A., Aranda R., Briere D. M., et al. The KRASG12C Inhibitor MRTX849 Provides In-sight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10(1): 54-71.
- [74] Hong D. S., Fakih M. G., Strickler J. H., Desai J., Durm G. A., Shapiro G.I., et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med. 2020, 383(13):1207-1217.
- [75] Briere D., Calinisan A., Aranda R., Sudhakar N., Hargis L., Gatto S., The KRASG12C Inhibitor MRTX849 Reconditions the Tumor Immune Microenvironment and Leads to Durable Complete Responses in Combination with Anti-PD-1 Therapy in a Syngeneic Mouse Model. AACR 2019. [Abstract]
- [76] Tran E., Robbins P.F., Lu Y.C., Prickett T.D., Gartner J.J., Jia L., et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med. 2016, 375(23):2255-2262.
- [77] Vakiani E., Janakiraman M., Shen R., Sinha R., Zeng Z., Shia J., et al. Comparative genomic analysis of primary versus meta-static colorectal carcinomas. J Clin Oncol. 2012, 30: 2956-2962.
- [78] Yoshimura A., Muto G. TGF-β function in immune suppression. Curr Top Microbiol Immunol. 2011, 350:127-47.
- [79] Boutin A. T., Liao W. T., Wang M., Hwang S. S., Karpinets T. V., Cheung H., et al. Oncogenic KRAS drives invasion and main-tains metastases in colorectal cancer. Genes Dev. 2017, 31:370-382.
- [80] Smeby J., Sveen A., Merok M. A., Danielssen S. A., Eilertsen I. A., Guren M. G., et al. CMS-dependent prognostic impact of KRAS and BRAFV600E mutations in primary colorectal cancer. Ann Oncol. 2018, 29:1227-1234.
- [81] Guinney J., Dienstmannn R., Wang X., deReyniès A., Schlicker A., Soneson C., et al. The consensus molecular subtypes of colo-rectal cancer. Nat Med. 2015, 21:1350-6.
- [82] Le D. T., Kim T. W., Van Cutsem E., Geva R., Jäger D., Hara H., et al. Phase II Open-Label Study of Pembrolizumab in Treat-ment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J Clin Oncol. 2020, 38:11-19.
- [83] Overman M. J., Lonardi S., Wong K. Y. M., Lenz H. J., Gelsomino F., Aglietta M., et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol. 2018, 36:773-779.
- [84] Andre T., Shiu K. K., Kim T. W., Jensen B. V., Jensen L. H., Punt C., et al. KEYNOTE-177: Phase 3, Open-label, randomized study of first-line pembrolizumab (Pembro) versus investigator-choice chemotherapy for mismatch repair-deficient (dMMR) or microsatellite instability-high (MSI-H) metastatic colorectal carcinoma (mCRC)[Abstract]. J Clin Oncol. 2018, 4-suppl (Feb-ruary 26th).
- [85] Ganesh K., Stadler Z. K., Cercek A., Mendelsohn R. B., Shia J., Segal N. H., et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol. 2019, 16:361-375.
- [86] Lal N., White B. S., Goussous G., Pickles O., Mason M. J., Beggs A. D., et al. KRAS Mutation and Consensus Molecular Subtypes 2 and 3 Are Independently Associated with Reduced Immune Infiltration and Reactivity in Colorectal Cancer. Clin Cancer Res. 2018, 24:224-233.
- [87] Liao W., Overman M. J., Boutin A. T., Shang X., Zhao D., Dey P., et al. KRAS-IRF2 Axis Drives Immune Suppression and Im-mune Therapy Resistance in Colorectal Cancer. Cancer Cell. 2019, 35:559-572.
- [88] Tran E., Ahmadzadeh M., Lu Y. C., Gros A., Turcotte S., Robbins P. F., et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015, 350:1387-90.
- [89] Yuki S., et al. Short-term results of VOLTAGE-A: Nivolumab monotherapy and subsequent radical surgery following preoper-ative chemoradiotherapy in patients with microsatellite stable and microsatellite instability-high locally advanced rectal can-cer. 2020 ASCO annual meeting. [Abstract 4100]
- [90] Ghiringhelli F., et al. Durvalumab and tremelimumab in combination with FOLFOX in patients with RAS-mutated, microsatel-lite-stable, previously untreated metastatic colorectal cancer (mCRC): Results of the first intermediate analysis of the phase Ib/II MEDETREME trial. J Clin Oncol. 2020 38: 15-suppl, 3006.
- [91] Shota F., et al. Regorafenib plus nivolumab in patients with advanced gastric (GC) or colorectal cancer (CRC): An open-label, dose-finding, and dose-expansion phase 1b trial (REGONIVO, EPOC1603). 2019 ASCO annual meeting. [Abstract]
- [92] Wang G., Wang J. J., Yin P. H., Xu K., Wang Y. Z., Shi F., et al. Strategies to target energy metabolism in consensus molecular subtype 3 along with Kirsten rat sarcoma viral oncogene homolog mutations for colorectal cancer therapy. J Cell Physiol. 2019, 234:5601-5612.
- [93] Yun J., Rago C., Cheong I., Pagliarini R., Angenendt P., Rajagopalan H., et al. Glucose deprivation contributes to the develop-ment of KRAS pathway mutations in tumor cells. Science. 2009, 325: 1555-9.
- [94] Kerr E. M., Gaude E., Turrell F. K., Frezza C., Martins C. P. Mutant KRAS copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature. 2016, 531: 110-3.
- [95] Gu M., Xu T., Chang P. KRAS/LKB1 and KRAS/TP53 co-mutations create divergent immune signatures in lung adenocarcino-mas. Ther Adv Med Oncol. 2020, ahead of print.