Parkinson’s disease (PD) is a progressive neurodegenerative disorder that arises due to a complex and variable interplay between elements including age, genetic, and environmental risk factors that manifest as the loss of dopaminergic neurons. Contemporary treatments for PD do not prevent or reverse the extent of neurodegeneration that is characteristic of this disorder and accordingly, there is a strong need to develop new approaches which address the underlying disease process and provide benefit to patients with this debilitating disorder. Mitochondrial dysfunction, oxidative damage, and inflammation have been implicated as pathophysiological mechanisms underlying the selective loss of dopaminergic neurons seen in PD.
- Parkinson’s disease
- neurodegeneration
- oxidative stress
- cyclic nitroxide
- mitochondrial dysfunction
- reactive oxygen species
- antioxidants
1. Introduction
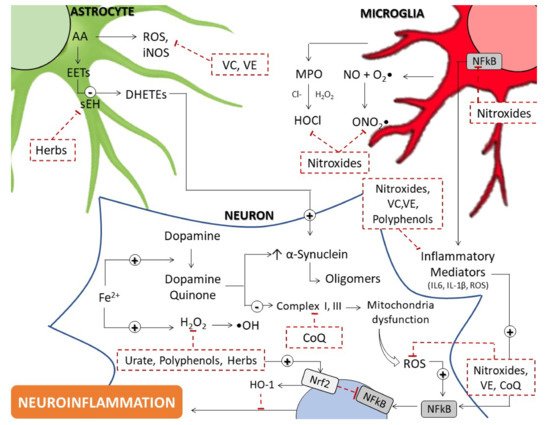
2. Evidence Supporting Enhanced Oxidative Stress in PD
2.1. Mitochondrial Dysfunction
2.2. Iron Dysregulation
2.3. Dopamine Quinone
3. Antioxidants
3.1. Endogenous Low-Molecular Weight Molecules
3.1.1. Coenzyme Q
Coenzyme Q10 (CoQ10) is a lipid-soluble antioxidant essential to mitochondrial electron transport chain (ETC) function and facilitates the transport of electrons from complex I and II to complex III [30]. As an antioxidant, CoQ10 directly reduces the initiation and propagation of lipid peroxidation by preventing lipid peroxyl radical production, as well as indirectly by regenerating other antioxidants such α-tocopherol (Vitamin E) and ascorbate (Vitamin C) [31][32][31,32]. CoQ10 is present in all intracellular membranes; however, as a component of the oxidative phosphorylation pathway, it is especially well placed in mitochondria, where electron leakage contributes to cellular superoxide and hydrogen peroxide production, to inhibit protein and DNA oxidation by both a general ROS scavenging mechanism as well as functioning as a chain-breaking antioxidant to prevent free radical propagation [32].
A significant deficiency of CoQ10 with functional ex vivo analysis has been shown in PD patients compared to age and gender-matched controls [33] as well as decreased total plasma reduced coenzyme Q10 (also referred to as ubiquinol10) and increased oxidized CoQ10, a marker of oxidative stress [34].
A neuroprotective effect of CoQ10 has been demonstrated in numerous in vitro PD studies. Thus, CoQ10 reduced oxidative stress and neuronal damage induced by paraquat in SHSY5Y cells [35][39] and has also been found to stabilize the mitochondrial membrane to reduce both total cellular and mitochondrial ROS generation and apoptosis induced by hydrogen peroxide in a neuronal cell model of PD [36][40]. The promising therapeutic potential of CoQ10 has been further established in in vivo studies with various PD models.
Clinical trials of CoQ10 have not replicated the promising results obtained in cell and animal models. Initial pilot studies and small clinical trials demonstrated that CoQ10 was safe and well-tolerated up to 1200 mg/d [37][38][44,45], and the minimal side effect profile of CoQ10 has also been substantiated in an unrelated study in Huntington disease as well as a more recent phase 3 RCT [39][40][46,47]. The Unified Parkinson’s Disease Rating Scale (UPDRS) is a clinical rating scale applied prior to 2008 in the assessment of PD severity. An early clinical trial with CoQ10 demonstrated a dose-dependent reduction in worsening of UPDRS; however, this study had a relatively small sample size of eighty enrolled patients, and its findings have not been reproduced in larger clinical trials [38][45]. The QE3 trial, the largest clinical trial of CoQ10 for PD to date, considered a participant population of six hundred and found that both 1200 and 2400 mg/d of CoQ10 demonstrated no clinical benefit, and in fact both treatment groups showed slight adverse trends in MDS-UPDRS scores relative to placebo [40][47]. The results of the QE3 trial have been substantiated by later meta-analyses, which concluded that current evidence indicates that CoQ10 does not slow functional decline, provide symptomatic benefit, or improved motor function [41][42][50,51].
Despite current evidence suggesting minimal symptomatic or disease-modifying effects of CoQ10, several promising novel therapeutic approaches have been identified, indicating that the potential clinical benefit of CoQ10 in PD has yet to be reached. The reduced form of CoQ10, which has been shown to have a better neuroprotective effect in animal models [43][53], was demonstrated to produce a statistically significant reduction in UPDRS scores in PD patients treated with a comparatively low dose of 300 mg/d as compared to the placebo [44][54]. Interestingly, the reduced form of CoQ10 tested did not elicit any clinical benefit in early PD patients who were not taking levodopa, suggesting that CoQ10 may be beneficial in specific PD patient populations, a nuance lost by studies with broader inclusion criteria [44][54]. Of note, a phase 2 clinical trial was registered in 2018 which will focus on employing an omics-based strategy to stratify PD patients into subgroups based on their genetic mitochondrial risk burden and their expected treatment response to CoQ10 [45][55]. The awaited results from this study could validate one of several promising novel directions for future research on the role of CoQ10 in the treatment of PD.
3.1.2. Urate
Urate is a prominent plasma antioxidant [46] [56] that inhibits lipid peroxidation and damage to biological molecules by acting as a free radical scavenger that neutralizes singlet oxygen, superoxide radical anion, and peroxynitrite (ONOO-) [47][48][57,58]. There is substantial epidemiological evidence that high urate levels are associated with a reduced risk for developing PD as well as a slower rate of clinical decline, although interestingly, despite robust evidence in men, this trend is variably observed in women, reflective of the complex interplay of factors influencing PD pathogenesis [49][50][51][59,60,61].
In differentiated PC12 cells, urate inhibited 6-OHDA induced neurotoxicity and reduced oxidative stress, as assessed by biomarkers such as lactate dehydrogenase (LDH), malondialdehyde (MDA), and 8-OHdG [52][62]. Furthermore, urate pretreatment in dopaminergic cells (SHSY5Y and MES23.5) attenuated 6-OHDA and hydrogen peroxide-induced cell death, and the mechanism underlying this neuroprotection was dependent on nuclear factor erythroid 2-related factor (Nrf2) activation and the upregulation of this transcription factor’s target antioxidant genes, -glutamate-cysteine ligase catalytic subunit (–GCLC) and heme oxygenase-1 (HO–1) [53][63]. Interestingly, urate and its precursor, inosine, were found to reduce the toxic effect of hydrogen peroxide as well as markers of free radical generation and oxidative damage in co-cultures of astrocytes and MES23.5 dopaminergic cells, but not in dopaminergic monocultures alone, suggesting that urate may provide multi-faceted neuroprotection in vivo [54][55][65,66]. Consistent with a neuroprotective mechanism identified in cell models, urate increased mRNA and protein expression of Nrf2 and its dependent antioxidant genes, -GCLC and HO–1, in mice treated with MPTP [56][67]. Clinical trials have been somewhat limited by the association of elevated urate and an increased risk of gout [57][69]. However, inosine, a urate precursor, has been found to be generally safe, well-tolerated, and effective in increasing serum and CSF urate levels as well as plasma antioxidant capacity [58][59][60][70,71,72]. The SURE-PD3 trial, a randomized, double-blind, placebo-controlled phase 3 trial of oral inosine treatment in two hundred and ninety-eight early PD patients closed early based on a pre-specified interim futility analysis [61][73]. This trial demonstrated no significant difference between inosine and the placebo in clinical progression or secondary efficacy outcomes such as dopamine transporter binding loss [61][73].
3.1.3. Glutathione
The glutathione (GSH) system is the main endogenous antioxidant defense and protects cell viability and function by a mechanism of free radical scavenging, interaction with iron metabolism, and transition metal chelation and as a cofactor for other antioxidants, such as glutathione peroxidase [62][74]. GSH also has a role in preventing mitochondrial dysfunction by maintaining the redox status of proteins via glutaredoxins, as well as the chemical reduction of toxic dopamine quinone formed during dopamine auto-oxidation [63][64][75,76]. Consistent with the role of oxidative stress in PD pathogenesis, there is a decrease in reduced glutathione and an increase in the levels of oxidized glutathione in post-mortem analysis of substantia nigra tissue [65][77]. N-acetylcysteine (NAC), a membrane-permeable cysteine precursor which functions as the rate-limiting substrate in GSH synthesis, is a potential therapeutic approach to target the GSH system, given that neurons cannot directly uptake GSH [66][80]. NAC has been found to ameliorate the decreased levels of GSH in the substantia nigra in a 6-OHDA hemi-parkinsonian rat model [67][81]. Similarly, intravenous infusion of NAC increases both brain GSH concentration and blood glutathione redox ratio in PD patients as well as healthy controls, although this effect is, as expected, more pronounced in the PD patient group [68][82]. However, the use of NAC in the treatment of PD is limited by a short half-life and poor oral bioavailability due to extensive first-pass metabolism [69][83]. Furthermore, scopoletin, a novel antioxidant moiety, restored redox balance and reduced oxidative damage and mitochondrial dysfunction in both MPP+ treated SH-SY5Y cells and a Drosophila genetic model of PD [70][85]. Interestingly, this mechanism of neuroprotection was mainly attributed to maintaining glutathione levels in its oxidizable form [69][83]. This suggests that the development of novel therapeutics which target the GSH system presents a promising approach to circumvent the pharmacological difficulties associated with administering GSH, or its precursor, NAC, while still harnessing the potent antioxidant capacity of this system. In clinical trials, a combination of intravenous infusions and oral doses of NAC was shown to significantly increase dopamine transporter binding in the caudate and putamen [71][86]. However, the unfavorable pharmacokinetics of oral NAC reduce the potential clinical utility of this approach. A prospective 4-week study found that although oral NAC increased peripheral antioxidants measures such as catalase and GSH/GSSG, this treatment failed to increase brain GSH or reduce markers of lipid peroxidation [72][87]. Furthermore, MDS-UPDRS scores increased in four of the five PD patients treated with oral NAC, a worsening of clinical parameters which the authors suggested may have occurred due to drug-drug interactions between NAC and other PD medications, such as amantadine [72][87].
3.2. Dietary Antioxidants
3.2.1. Vitamin C and Vitamin E
Vitamin C (ascorbate) is a water-soluble vitamin that functions as a co-factor in several biosynthesis pathways, including collagen, catecholamine, and neuropeptide production, as well as maintenance of redox homeostasis as a prominent antioxidant [73][89]. Ascorbate acts as a free-radical scavenger and antioxidant through the non-enzymatic reduction of ROS such as superoxide, hydroxyl, and peroxyl radicals [74][90]. Ascorbate also functions in the regeneration of Vitamin E, another antioxidant, by reducing the tocopheroxyl radical [74][90].
Cell and animal studies of vitamin C support the therapeutic potential of this antioxidant in the treatment of PD. In a model of dUCH using Drosophila, where a homolog of the PD-related gene UCH-L1 was a knockdown, treatment with low dose ascorbate reduced dopaminergic neuron loss and motor dysfunction, although side effects on physiology occurred with high doses and long-term treatment [75][94]. Ascorbate also reduces ROS and inducible nitric oxide synthase (iNOS) in MPP+ treated astrocytes as well as upregulates endogenous antioxidants through a mechanism of NFκB activation in both astrocytes and MPTP treated mice [76][95]. The use of novel adjunct agents is a promising direction of research to improve the efficacy of vitamin C. A meta-analysis of twelve studies, which considered dietary intake of vitamin C, concluded that there was no significant reduction in risk of developing PD in the high vitamin C intake group as compared to the low intake group [77][97]. Furthermore, six of the studies included in this meta-analysis also found that supplemental vitamin intake did not have any effect on the development of PD [77][97]. However, ascorbate supplementation has also been shown to positively impact levodopa pharmacokinetics, improving both absorption and bioavailability, suggesting that the therapeutic role of ascorbate is not limited to reducing PD development [78][98]. On the other hand, Vitamin E (α-tocopherol) is the major lipophilic antioxidant in the brain. Chronic vitamin E deficiency in this organ coincides with lipid peroxidation, altered phospholipid composition, and energy metabolism together with impaired cognition in a recognized model of brain impairment [79][99]. In addition, α-tocopherol has been proposed as a therapeutic target in PD and other neurodegenerative diseases, as deficiency or malabsorption cause neurological syndromes such as ataxia and nerve damage. Low blood and CSF vitamin E concentrations are consistently observed in neurodegenerative disease [80][101] along with oxidative stress [81][102], and supplementation improves cognitive decline and reduces lipid peroxidation and amyloid deposition [82][103]. Vitamin E elicits beneficial effects in animal studies of PD. For example, supplementation slows dopaminergic neuron loss in a model of superoxide perturbation [83][104] and attenuates both behavioral and biochemical abnormalities after 6–OHDA challenge in rats [84][105]. Chronic vitamin E also restored synaptic plasticity in PINK–1 K/O mice, a model of subclinical PD that shows decreased dopamine release in the absence of overt neuron loss [85][110]. This study suggested that neuroprotection resulted from vitamin E-induced improved mitochondrial bioenergetics like that observed with a water-soluble vitamin E analog (Trolox) and mitochondrial complex 1 and IV activity in PINK–1-deficient dopaminergic cells [86][111]. Importantly, vitamin E also shows efficacy in improving T-cell function and immune responses in aged human studies [87][113]. These positive outcomes may be related to antioxidant activities that maintain bio-membranes and, therefore, effective signaling that is imperative for correct immune responses and/or via gate-keeping levels of lipid peroxidation products that act as inflammation mediators. Both animal and human studies show vitamin E supplementation reduces the proinflammatory mediator, prostaglandin E2 and other pro-inflammatory cytokines such as TNF-α and IL–6 [88][112]. Dietary vitamin E insufficiency is associated with PD in case-control studies [89][116]; however, when measured, low vitamin E concentrations have been reported in only some [90][91][117,118] and not all [80][92][93][94][101,119,120,121] human PD studies. A long-term follow-up of high dose α–tocopherol in early disease patients also failed to demonstrate positive outcomes on mortality [95][96][125,126]. Overall, despite positive preclinical studies of neuron loss, inflammation, and oxidation, clinical trials of vitamin E to date have not demonstrated benefit on cognition, neurodegeneration, or mortality in PD. Other vitamin E forms have been tested in neurodegeneration such as tocotrienols (T3s; from palm oil), which have lower bioavailability than α-tocopherol but exert antioxidant and other bioactivities such as mediating cell signaling and are accessible to the brain/CNS. This suggests that bioactive vitamin E analogs with antioxidative and anti-inflammatory activities may be neuroprotective and should be tested further for efficacy in PD. Tocotrienol-rich vitamin E (Tocovid) possesses potent antioxidant and anti-inflammatory properties and is currently in clinical trials for PD (NCT04491383) [97][129].
3.2.3. Polyphenols and Flavonoids
Polyphenols are a large group of chemically reactive plant micronutrients credited with the health benefits of fruit/vegetable-rich diets and can be divided into subclasses; phenolic acids, flavonoids, lignans, and stilbenes, of which flavonoids comprise > 60%. Polyphenols demonstrate antioxidant and metal chelating activities in vitro [98][130] suggesting an ability to moderate neurodegeneration and oxidative processes in the brain. Dietary polyphenols are associated with reduced risk of dementia [99][133], cognitive decline [100][134], and PD [101][135]. PD locomotor symptoms are significantly delayed by drinking tea containing various flavonoids in a retrospective study [102][136] and reduction in PD risk was also determined in meta-analyses of tea consumption in case-controlled [103][137] and prospective cohort’s studies [104][138]. Neuroprotection with polyphenol supplementation via anti-inflammatory, anti-apoptotic, and antioxidant activities has been demonstrated in numerous cell and animal PD models [105][140]. Despite this growing body of literature, the precise mechanism/s whereby polyphenols may be neuroprotective are not well defined. Direct antioxidant scavenging of cellular oxidants may be unlikely given the low bioavailability of polyphenols and limited ability to cross BBB, and moreover, that endogenous antioxidants likely outcompete kinetically. Indirect antioxidant activities in cells include activation of the Nrf2/antioxidant response element (ARE) pathway that regulates phase 2 antioxidant and detoxification enzyme expression [106][107][142,143]. Polyphenols are also low affinity ligands for various cell signal kinases involved in inflammation, mitochondrial respiration, and apoptosis such as AMPK, PI3K/AKT/mTOR pathway, p38 MAPK, and ERK1/2 [108][144] and kinase dysfunction is genetically linked to PD and thought responsible for hyperphosphorylation of _α–synuclein [109][145]. Antioxidant activities of polyphenols include ROS reduction, maintenance of endogenous antioxidants and mitochondrial function, and a tight interaction between polyphenol antioxidant activity and locomotor function is found in various PD models [110][146]. For example, green tea polyphenols are neuroprotective in 6-OHDA model via inhibition of ROS, NO, lipid peroxidation, and iNOS [111][147]. Quercetin reduces lipid peroxidation and restores SOD and CAT enzymic activity and GPx/GSH across rodent PD models [112][150] and increases Na+/K+ATPase and mitochondrial complex I activity. Epigallocatechin-3-gallate (EGCG), an abundant tea catechol, reduces lipid peroxidation and apoptosis and improves locomotor function in a PD fly model [113][153]. EGCG also regulates iron export and reduces oxidative stress and protein carbonyl formation in MPTP-treated mice [114][154] to moderate neuron injury and neuroinflammation. In addition, other polyphenols have been described as potential compounds in the treatment of PD. Naringenin increases Nrf2, activates ARE genes, and is neuroprotective after 6-OHDA treatment in vitro and in vivo [115][156]. Baicalein suppresses NFkB translocation, and this is accompanied by decreased kinase activity and the promotion of neurogenesis and neurotrophin signaling [116][158]. Individual polyphenols exhibiting multifactorial antioxidant, anti-inflammatory, and anti-apoptotic effects that modulate protein aggregation, ROS, mitochondrial dysfunction, and neuroinflammation to protect neurons and reverse pathological features have been investigated in PD. Resveratrol has not been tested in PD clinical trials, but two trials in AD (which demonstrates common metabolic dysfunctions and pathogenesis) have shown safe use of resveratrol and penetration of BBB to elicit CNS effects. Thus, decreased CSF MMP–9 degrades extracellular matrix and may protect the CNS [117][169], causing a slower decline in β-amyloid peptide, indicating reduced accumulation in the brain [117][118][169,170]. Curcumin increases GSH in the brain and decreases protein oxidation while maintaining complex I activity [119][148] and SOD1 [120][149]. Both curcumin and resveratrol show low absorption and oral bioavailability [121][175] and this may affect therapeutic potential particularly in neurodegeneration. Multifactorial and off-target effects of polyphenols may be a concern, and the metabolism of these natural products can also further limit their bioactivity and bioavailability. Further investigations that show significant polyphenol delivery to plasma/CSF with functional effects in CNS will be important.
3.3. Bioactive Natural Herbal Extracts
The use of natural herbs in the treatment of neurodegenerative diseases has been widely recognized by Traditional Chinese Medicine and is often associated with few (or low incidence of) adverse effects in humans. The presence of polyphenols and related phytochemicals with biologically active properties in these herbal preparations is increasingly a focus in contemporary research, especially in the field of developing new complementary therapies [122][177] that may provide therapeutic benefits. For example, Kurarinone, a major constituent of the Sophorae Flavescentis Radix, (referred to as Kushen in China), is commonly used to treat diarrhea and inflammation-related disorders [123][178]. Additionally, 4′,Polydatin (3,4′,5-trihydroxystilbene-3-β-D glucoside), a natural resveratrol glucoside extracted from Polygonum cuspidatum, exhibits pharmacological effects against degenerative motor function by protecting dopaminergic neurons in the rat substantia nigra [123][178]; here polydatin decreased microglial activation through stimulating the AKT/GSK3β-Nrf2 signaling axis to ameliorate lipopolysaccharide-mediated experimental PD [124][179] indicative of an anti-inflammatory action for polydatin.
Current pharmacological research indicates that Astragaloside IV (AS-IV), an antioxidant derivative of Astragalus membraneaceus Bunge, can both relieve inflammation and improve behavioral disorders in an MPTP-induced mouse model of PD [125][180]. Furthermore, a substantial body of evidence indicates that herbal medicines can attenuate neuroinflammation in PD through diminishing iNOS-dependent nitric oxide release (thereby down-regulating the formation of damaging ONOO-), COX–2 activity in glial cells, unregulated ROS generation, and decreasing the expression of inflammatory cytokines such as TNF-α and IL–6 [123][126][127][178,184,185].
Studies have described herbal tonics that prevent dopaminergic neurodegeneration through the suppression of microglia activation [124][179] and inhibition of astrocyte senescence [124][179]. Importantly, several studies have indicated that these protective activities involve a common suppression of NFκB signaling pathways. In LPS-induced mice, the administration of 10 mg/kg of Icariside II, an active flavonoid extracted from Epimedium, diminishes inflammation by increasing IκB–α protein accumulation in the hippocampal tissues, ultimately ameliorating NFκB transcriptional activation as judged by monitoring tissue levels of NFκB-p65 phosphorylation [128][186].
Alternate mechanisms of action include neuroprotection offered by traditional medicinal plants through enhancing endogenous antioxidant enzymes systems such as SKN–1/Nrf2 activation via the MAPK pathway in a Caenorhabditis elegans model of PD [129][188]. More recent studies have implicated a key role of soluble epoxide hydrolase (sEH) enzyme and its biological substrates such as eicosatrienoic (EETs) and epoxydocosapentaenoic (EDPs) acids in the regulation of neuroinflammation [130][189]. Thus, pharmacological inhibition of sEH (or through sEH gene deletion) protects the mouse brain from MPTP-mediated neurotoxicity; notably, a positive correlation between sEH expression and the extent of α-synuclein phosphorylation was identified in the striatum tissue in this experimental model of PD. In strong support for this pathway in humans, patients with Lewy body-dementia and other chronic psychiatric diseases showed elevated sEH protein in post-mortem brains [131][132][190,191], further implicating sEH in the pathogenesis of neuroinflammation that is characteristic of PD. In the same MPTP-induced PD mice model, the traditional Chinese herbal medicine Sophora flavescens alleviated dopaminergic neurotoxicity including loss of neurotransmitters and expression of TH in the substantia nigra and striatum and this resulted in an improved gait. Interestingly, kurarinone, the active constituent of Sophora flavescens, showed an inhibitory effect against sEH by interacting with amino acid residues, stimulating an increase in plasma levels of EETs and a corresponding decrease in its oxidation product(s) such as dihydroxyeicosatrienoic acids [123][178].
The main clinical trials cited and other most recent clinical trials have been collated in a tabulate and this information can be found at Leathem A. et.al., 2022 (https://www.mdpi.com/1422-0067/23/13/6923)