The neuroprotective effects of HupA treatment result from cholinergic signaling, which increases Wnt signaling, nonamyloidogenic processing of the amyloid precursor protein (APP), neurotrophic factor expression, synaptic activity, antagonism of the N-methyl-D-aspartate (NMDA) receptor (NMDAR), modulation of ROS, iron homeostasis and neuronal survival.
- neurodegenerative diseases
- huperzine
- AChEI
- Alzheimer’s disease
- therapeutic potential
- neuroinflammation
1. Introduction
Its IUPAC name is (1R,9S,13E)-1-amino-13-ethylidene-11-methyl-6-azatricyclo[7.3.1.02,7]trideca-2(7),3,10-trien-5-one. With a molecular weight of 242.32 g/mol [1][2][9,10], HupA presents a complex array of structural components: a compact tricyclic structure with a bicyclo [3.1.1] skeleton fused with an α-pyridone ring, an exocyclic ethylidene moiety, and a 3-carbon bridge with an -NH 2 group. This complex and unique molecular structure accounts for the interesting biological functions and the high stability of HupA [3][11], plus it results in the presentation of the correct electrostatic field for binding to AChE [4][5][12,13]. The three-dimensional structure of AChE complexed with the nootropic alkaloid [-]-HupA was resolved at the Weizmann Institute of Science, Israel [6][14]. Notably, chemically synthesized [−]-HupA inhibits AChE with similar effectiveness as the natural molecule [2][10], and [+] -HupA ’s AChE inhibitor (AChEI) activity is at least 50-fold less potent than that of the naturally occurring stereoisomer [7][15].
The enzyme AChE catalyzes the hydrolysis of the neurotransmitter acetylcholine into choline and acetic acid, regulating the return of cholinergic neurons to a resting state [8][16]. Most of the cortical AChE activity present in the brain of AD patients is known to be predominantly associated with the amyloid core of senile plaques [9][10][17,18]. The globular tetramer (G4) of AChE present in the central nervous system (CNS) [11][19] (see Figure 1 ), forms a stable toxic complex with the amyloid-β (Aβ) peptide during its assembly into Aβ filaments and increases the aggregation and neurotoxicity of amyloid fibrils [12][13][14][20,21,22].
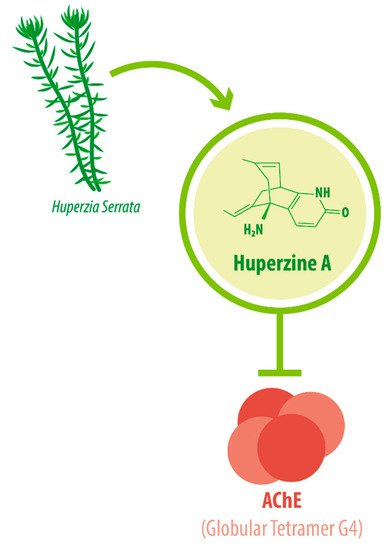
Due to its potent AChEI activity, HupA is used as a treatment for AD. According to the World Health Organization (WHO), 135.5 million people worldwide will be living with dementia by the year 2050. AD accounts for 65% of all dementia cases [15][24]. It is a progressive neurological degenerative disorder affecting memory, among other cognitive functions [16][17][18][25,26,27]. Vascular-associated dementia (VaD), on the other hand, is caused by various cerebrovascular diseases, such as cerebral infarction and hemorrhage [19][28]. These conditions impose great monetary and emotional costs on patients and their caregivers. HupA is approved as the drug of choice for Alzheimer’s disease (AD) treatment in China and as a dietary supplement in the United States [20][21][29,30], as intake can improve AD pathogenesis [22][31].
The biological functions of HupA have been studied both in vitro and in vivo. The present review aims to provide an integrated perspective of the neuroprotective molecular signaling of HupA in dementia, especially in AD.
2. HupA within the CNS: Modulation of Critical Pathological Conditions
One of the hallmarks of AD is the accumulation of Aβ peptide (both oligomers and fibrils) that associate with AChE to form amyloid plaque, producing neuronal cell death [12][13][20,21]. The neuroprotective effect of HupA is in part mediated by a dose-dependent reduction in subcellular amyloid-β accumulation in the cortex and hippocampus as shown in a double transgenic mice model carrying the Swedish-mutant amyloid precursor protein (APP) and a deletion in exon 9 of presenilin 1 (PS1) (known as APPswe/PS1 mice) [23][32], although it is noteworthy that this effect is reportedly absent in TgCRND8 mice [24][33]. These reductions occur at the protein level through a dose-dependent modulation of the activity of β-site of the APP-cleaving enzyme (BACE1) activity, a reduction in PS1 expression, and a marked increase in α -secretase cleavage, promoting the non-amyloidogenic processing of APP [23][25][32,34]. An improvement in spontaneous working memory of 68% has been observed in APPswe/PS1 mice after HupA treatment, although HupA does not seem to improve cognition in wild-type (WT) mice [26][35]. In primary cortical neuron cultures, HupA ameliorates oligomeric Aβ42-induced neurotoxicity by reducing the intracellular accumulation of Aβ42 [27][36].
At the same time, HupA treatment attenuates the Aβ load in brain mitochondrial homogenates and ameliorates the mitochondrial swelling observed in APPswe/PS1 mice, although it has no effect on WT mice [26][35]. It has been reported that Aβ accumulation causes mitochondrial dysfunction, resulting in neurotoxicity. HupA treatment restores adenosine triphosphate (ATP) levels and reduces reactive oxygen species (ROS) levels increased by Aβ42 in neurons, and prevents Aβ42-mediated destabilization of the mitochondrial membrane [27][36]. Notably, HupA treatment reduces Aβ42 accumulation in mitochondria-enriched cellular fractions, but has no significant effects on membrane or cytosol Aβ42 levels, indicating that it exerts a specifically mitochondrial effect [27][36].
HupA also ameliorates Aβ25–35-induced apoptosis. A proteomic analysis showed that HupA downregulates 29 proteins, including the tumor suppressor protein p53, which has a direct link with apoptosis [28][37]. In a co-culture system of neural stem cells and microglia exposed to Aβ1–42, HupA treatment partially reduced the secretion of inflammatory factors interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), and macrophage inflammatory protein-1α (MIP-1α) [28][37]. HupA also significantly increases the ratio between B-cell lymphoma-2 (Bcl-2) and Bcl-2-like protein 4 (Bax), resulting in improved cell viability. Similarly, HupA directly acts on microglial cells to reduce the expression of cytokines and chemokines [29][38]. In primary astrocyte cultures, HupA preincubation reduces Aβ1-42-induced cell damage, preventing peaks in the release of p65 subunit of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) [30][39].
Several studies have evaluated the potential benefits of HupA in human patients. HupA treatment in human subjects suffering from dementia (AD o VaD) shows evidence of improved cognition [31][19][32][33][4,28,40,41]. It has also been reported that eight weeks of HupA treatment for AD patients improved task switching and alleviated cognitive impairment [34][42]. The benefits of HupA treatment are apparent when using measurement tools such as Mini-Mental State Exam (MMSE), the AD Assessment Scale-cognitive subscale (ADAS-Cog)) or the ADAS-noncognitive subscale (non-COG) tests. Measuring with tools such as Hastgawa Dementia Scale (HDS) or Wechsler Memory Scale (WMS) tests showed no significant improvement from HupA treatment [15][16][24,25]. These results were corroborated in a randomized clinical trial in which HupA treatment in VaD patients significantly improved cognitive function according to the MMSE and Activities of Daily Living (ADL) tests but not according to the Clinical Dementia Rating (CDR) scores [15][19][24,28]. However, other trials conclude that HupA treatment could be more beneficial than psychotherapy and conventional medicine for VaD patients [31][4]. A small trial testing the effects of HupA tablets in AD patients showed that the intake of 0.2 mg of HupA improved cognition and memory in 58% of patients with no severe side effects [35][43], and a phase II multicenter, 3-arm randomized, double-blind placebo-controlled trial, evaluated as having high methodological quality in later reviews [31][4], showed significant cognitive enhancement in patients receiving 0.4 mg of HupA twice a day. Remarkably, this dose was well tolerated for 24 weeks even though most AD patients reported being unable to tolerate currently marketed AChEIs for a long period of time [32][36][40,44]. A more recent analysis concludes that HupA improves cognition in AD patients, and that its effects are dose and duration-dependent [37][45].
3. Neuroprotective Molecular Signaling of HupA against AD
The neuroprotective effects of HupA treatment result from cholinergic signaling, which increases neurotrophic factor expression, synaptic activity, antagonism of the N-methyl-D-aspartate (NMDA) receptor (NMDAR), modulation of ROS, and neuronal survival [38][1][3,9].
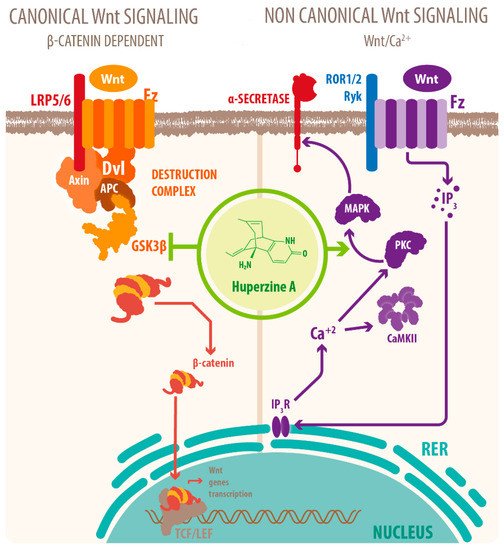
Activation of mAChRs and protein kinase C (PKC)/MAPK pathways have been proposed as a mechanism for this effect [7][39][40][15,23,46], as well as HupA mediated increase of phosphorylation of ERK1/2, that can also regulate ADAM10 transcription. Another mechanism for the cholinergic modulation of APP processing is the activation of Wnt signaling. Wnt signaling plays a crucial role in presynaptic assembly, postsynaptic differentiation at vertebrate peripheral neuromuscular synapses, stem cell differentiation, and the development and maintenance of the CNS [41][42][43][63,64,65]. Recent evidence suggests that it also regulates vascular stability, BBB integrity, and inflammation [44][66]. In the canonical Wnt pathway or Wnt/β-catenin pathway, Wnt ligands bind to receptors of the Frizzled (FZD) and low-density lipoprotein-related protein (LRP) families on the cell surface and activate several cytoplasmic relay components, including GSK-3β and adenomatous polyposis coli (APC), finally signaling to β-catenin. β-catenin enters the nucleus and forms a complex with lymphoid enhancer-binding factor 1 (LEF1) to activate the transcription of Wnt target genes. Wnt signaling pathways that are independent of β-catenin are referred to as non-canonical pathways, which include the planar cell polarity (PCP) pathway, also known as the Wnt/Jun N-terminal kinase (JNK) pathway, and the Wnt/ Ca 2+ pathway [41][63] (see Figure 24 ).
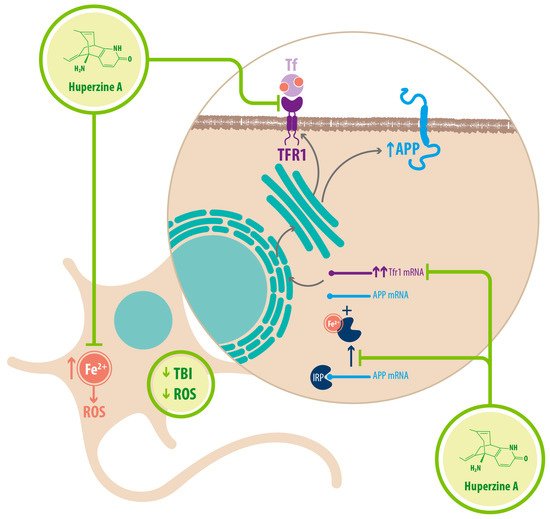
Regarding Fe 2+ homeostasis, it has been reported that AD mice models have elevated levels of Fe 2+ in the cortex and hippocampus compared with WT mice. Accordingly, the expression of transferrin receptor protein 1 (TFR1) is upregulated in APPswe/PS1dE9 mice, which explains the increase in Fe 2+ uptake [25][34]. Fe 2+ -mediated neurodegeneration might originate in the downregulation of α-secretase activity, inhibiting the nonamyloidogenic pathway, with concomitant increase of APP expression due to a functional iron-responsive element (IRE) that has been identified in the APP promoter, which would explain why APP levels increase after Fe 2+ treatment and decrease after chelator treatment. An alternative explanation could be that the translational expression of APP could be directed via release of a classic repressor interaction between APP mRNA and iron regulatory protein-1 (IRP-1) [25][34]. HupA might act directly as an Fe 2+ chelator, reducing the capacity of IRP-1 to induce APP translation [25][34]. HupA also downregulates TFR1 expression in mice in vivo, which reduces the uptake of transferrin-bound iron (TBI) in neurons [25][34] (see Figure 36 ) (see Table 12 ).
| Mechanism | Molecular Target | Effects of HupA Treatment | References |
|---|---|---|---|
| Mitochondrial protection | Upregulation of oxidative phosphorylation proteins | Recovered energetic homeostasis | [45] |
| Inhibition of cytoplasmic translocation of CytoC | Prevented mitochondria-mediated apoptosis | [46] | |
| Downregulation of AIF | Prevented cas3-independent apoptosis | [45] | |
| Inhibition of Caspase-3 cleavage | Prevented cas-3 dependent apoptosis | [45] | |
| Upregulation of GSH-PX SOD CAT |
Decreased ROS accumulation Decreased oxidative stress |
[45] | |
| Fe2+ homeostasis | Chelator/ Reducer of Fe2+ | Increased nonamyloidogenic processing of APP Downregulated expression of APP through IRP-1 |
[25] |
| Downregulation of TFR1 | Decreased Fe2+ uptake in neurons | [25] |
Finally, it is noteworthy that HupA reduces proinflammatory cytokines expression through activation of nAChRs. Activating the cholinergic anti-inflammatory pathway, the efferent arc of the inflammatory reflex in the CNS, results in suppressed inflammatory cytokine expression and systemic inflammatory responses. The activation of α7nAChR downregulates macrophages, microglia, and astrocytes activation inhibiting the production of proinflammatory cytokines while anti-inflammatory cytokine expression is not altered [2][40][47][10,46,71]. The cholinergic anti-inflammatory effects of HupA downregulates IL-1β, IL-6, and TNF-α secretion and NF-kB signaling, attenuating neuroinflammatory response [48][72].
4. Concluding Remarks
HupA has therapeutic benefits for the treatment of AD, but the understanding of molecular interactions involved in the recovery of neuronal function after HupA treatment is still incomplete. What is known is that AChEI activity is crucial through the regulation of Aβ peptide accumulation by activation of α-secretase cleavage and down-regulation of β/γ-secretase, enhancing BDNF/TrkB signaling as well as PI3K/Akt and PI3K /TrkB/mTOR pathways. Concomitant reduction of IL-1β, IL-6, TNF-α, and NF-kB signaling preserves neuronal function. Modulation of Wnt signaling through HupA treatment might be the most promising therapeutic target, as Wnt is involved in neuronal survival and in synaptic plasticity. There are also non-cholinergic neuroprotective effects such as the preservation of mitochondrial structure and function under Aβ insult, as well as the recovery of Fe 2+ homeostasis in the brain. Further research and development of HupA as a common treatment might alleviate the burden related to highly prevalent conditions that are costly in terms of public health policies and devastating to those who bear them and their caretakers.