Plectin is a multi-faceted, 500 kDa-large protein, which due to its expression in different isoforms and distinct organs acts diversely as a cytoskeletal crosslinker and signaling scaffold. It functions as a mediator of keratinocyte mechanical stability in the skin, primarily through linking intermediate filaments to hemidesmosomes. Skin fragility may occur through the presence of mutations in the gene encoding for plectin, PLEC, or through the presence of autoantibodies against the molecule.
- epidermolysis bullosa
- paraneoplastic pemphigus
- hemidesmosome
- intermediate filaments
- plakin
- skin blistering
- muscular dystrophy
- pyloric atresia
1. Introduction
Plectin, a member of the plakin family, is a 500 kDa-large protein with various roles as a cytoskeletal crosslinker and signaling scaffold. Twelve isoforms of the molecule have been described, arising through alternative splicing, primarily within the N-terminal domain [1]. Plectin is a multi-faceted, highly interesting molecule since different isotypes are expressed in various cells (including keratinocytes, fibroblasts, myoblasts, and Schwann cells), fulfilling distinct functions [1]. Only four are expressed in the epidermis, namely P1, P1a, P1c, and P1f [1]. Structurally, plectin has a central α-helical coiled-coil rod domain with N- and C-terminal globular domains [2]; its role in the skin and other organs is revealed by the effects of mutations in the distinct plectin domains and isoforms [3]. In the skin, plectin is a mediator of keratinocyte mechanical stability, primarily through the connection of intermediate filaments (IFs) to hemidesmosomes (HD) [4]. Specifically, the N-terminus contains an actin-binding domain and a plakin domain with binding sites for integrin α6β4 [5][6][5,6] and collagen XVII/ BP180 [7], while the C-terminus comprises six plakin domains with binding sites for IFs, vinculin, and integrins [8][9][10][8,9,10]. Plectin is also a component of desmosomes and focal adhesions [11]. It has been suggested that the binding of plectin to integrin β4 prevails over that with the actin cytoskeleton, resulting in the preferential distribution of plectin in the hemidesmosomes [12], which accounts for the important role of plectin in regulating keratinocyte adhesion. It also plays a role in organizing mitochondria and nuclei within the intermediate filament network [11][13][11,13], thus being involved in the cellular metabolome [14]. Skin fragility may occur through the presence of mutations in the gene encoding for plectin, PLEC , or through the presence of autoantibodies directed against the molecule. To date, more than 100 mutations have been reported to result in different inherited disease subtypes (Human Gene Mutation Database- HGMD 2021.1). The manifestations of these specific disorders are summarized below.
2. Plectin in Hereditary Skin Fragility Disorders
The PLEC gene is a huge gene on chromosome 8q24, consisting of 32 exons. Mutations have mostly been disclosed within exons 31 and 32 and cause the skin fragility disorder epidermolysis bullosa simplex (EBS) ( Figure 1 A–C). On the tissue level, plectin deficiency results in a split within the keratinocytes of the basal layer of the epidermis, and although considered exceedingly rare, around 8% of patients with EBS carry mutations in the PLEC gene [15]. Since blisters are within the basal keratinocytes directly above the HD, only fragments of the destroyed HD exist on the blister floor, and thus old blisters frequently appear to be junctional (also called “pseudojunctional” EBS) ( Figure 1 D,E) [16][17][16,17]. Electron microscopy reveals hypoplastic HD, with normal extracellular portions but a poor inner plaque assembly [16][18][16,18]. A variety of antibodies have been described to specifically detect different plectin domains, which has proven helpful for the prognostication of patients’ disease severity, as explained below.
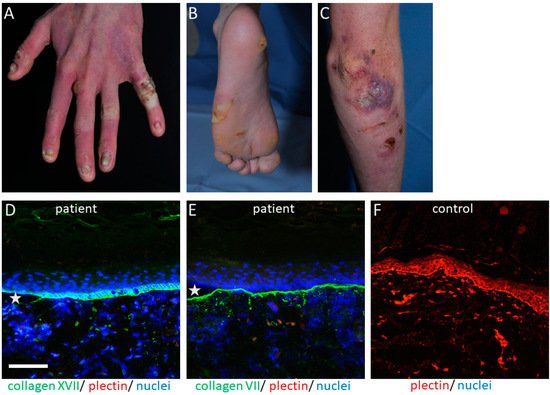
The clinical hallmark of the disease is the life-long, trauma-induced blistering of skin and mucosa. The EBS-PA subtype is characterized by congenital pyloric atresia and severe skin fragility, with areas of aplasia cutis, and was originally thought to be associated with an early demise [19][25]. However, a few cases have been reported where the patients survived the neonatal period, most of them developing muscular dystrophy and, interestingly, urological abnormalities [20][21][22][22,26,27].
Exceedingly rarely, patients might present myasthenic symptoms [23][24][25][26][37,38,39,40], although these symptoms might be disregarded in already severely muscularly impaired patients [25][39]. To date, phenotype–genotype correlations are still missing for the development of myasthenia in EBS-MD-affected individuals.
3. Plectin in Autoimmune Skin Fragility Disorders
Autoantibodies against plakins have been associated with autoimmune blistering disorders, specifically with paraneoplastic pemphigus (PNP), a life-threatening and rare disease, but also cases of more common bullous pemphigoid (BP) [27][28][47,48]. Due to its cytoplasmatic localization, pathogenicity, and development of anti-plectin antibodies, it is not fully understood, and the detection of plectin by blotting or immunoprecipitation is difficult due to its large size of 500 kDa [3]. Anti-plectin reactivity in BP, an acquired subepidermal blistering disease with autoreactivity to mainly hemidesmosomal proteins BP180 and/or BP230 [29][49], is a rare phenomenon and only rarely reported, while it is considered to occur through epitope spreading [30][31][32][50,51,52]. An analysis of plectin subdomains suggested the rod domain as being the immunodominant epitope [28][48].
Clinical hallmarks of PNP are polymorphous erosions, crusts, blisters, and/or lichenoid lesions with chronic and treatment-refractory mucositis, cheilitis, and conjunctivitis [30][50]. In addition, the disease shows an association with solid tumors or hematologic neoplasms (especially stomach, lung, or colon cancer, thymomas, and B-cell lymphomas) [33][53]. In 2001, the systemic nature of PNP was discussed by characterizing the multiorgan involvement occurring in patients, including respiratory or muscular manifestations, and the term “paraneoplastic autoimmune multiorgan syndrome” was introduced [31][51]. Direct immunofluorescence shows epithelial cell surface depositions of IgG and C3, but in around 50% of cases linear C3 also exists along the basement membrane zone of patients’ skin [32][52]. Indirect immunofluorescence on a monkey esophagus reveals IgG cell surface deposition, as occurring in pemphigus vulgaris or pemphigus foliaceus [30][50]. Several antigens have been associated with PNP. In the majority of patients, these include, besides plectin, the plakin family member envoplakin, desmoplakin I and II, desmogleins, desmocollins, BP230, and anti-alpha-2-macroglobulin -like 1 [34][35][36][37][54,55,56,57].
4. The Role of Plectin in Fibroblast Organization
The role of plectin in skin fibroblasts and its relevance for cutaneous disorders has remained largely unexplored. Plectin is connected to vimentin and plays a role in its network distribution within fibroblasts during the early stages of filament assembly [4]. It affects the mitosis, cell shape, polarization, and size of the cells. It also participates in forming connections between fibroblasts and their underlying extracellular matrix, while plectin-deficient fibroblasts have fewer protrusions and are rounder [38][58]. Their mechanical properties also seem to be impaired [39][59], since they are stiffer, seem to migrate less [40][44], and are more susceptible to stress-induced disruption [41][60].