Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Satoshi Nagayama and Version 2 by Rita Xu.
In the field of colorectal cancer (CRC) treatment, diagnostic modalities and chemotherapy regimens have progressed remarkably in the last two decades. However, it is still difficult to identify minimal residual disease (MRD) necessary for early detection of recurrence/relapse of tumors and to select and provide appropriate drugs timely before a tumor becomes multi-drug-resistant and more aggressive.
- liquid biopsy
- minimal residual disease
- cancer precision medicine
- immune checkpoint inhibitor
- neoantigen
1. Introduction
In spite of remarkable progress in diagnosis and treatment of colorectal cancer (CRC), there are still big challenges to improve the overall prognosis. Current diagnosis of recurrence/relapse is based on tumor biomarkers or imaging modalities including CT, MR, and PET examinations, which fail to detect minute lesions (micrometastases). It should be favorable to know the precise condition of the disease earlier than imaging diagnosis and initiate a proper treatment before clinically overt recurrences are identified. In addition, most patients who receive systemic chemotherapy become resistant during the treatment course and end up in the termination of the treatment with the standardized guidelines. A suitable drug should be selected and provided based on the molecular biological profiles of the tumors before the disease culminates in a far-advanced stage.
With the advance of genomic sequencing technologies typified by next-generation sequencing (NGS), it becomes much easier, more rapid, and less expensive to access comprehensive genomic information of the tumors. There is a high possibility that usage of the in-depth genomic profiles contributes to detection of recurrent/relapsed tumors and to proper choice of beneficial treatment options, including molecularly targeted therapies. In order to obtain the genomic profiles, blood-based tests, so called ‘liquid biopsies’, are considered to be a more useful method as compared to tumor biopsies, since liquid biopsies can be performed repeatedly and less invasively during a monitoring period and can provide the genomic information that is highly concordant with that of tumor biopsies. Hence, by leveraging the genomic profiles obtained by liquid biopsies, recurrence/relapse can be detected earlier than the current imaging diagnosis, and more suitable therapies can be provided without the delay in the time course of the treatment. Furthermore, analyzing the comprehensive genomic information, which also can be supplemented by liquid biopsies, will lead to a new development of effective immunotherapy related to mutations and/or neoantigens. In this review, we focus on potential roles of liquid biopsy in terms of clinical management of CRCs ranging from early detection of recurrence/relapse to acquisition of definitive clues leading to a promising and beneficial treatment, and touch on the potential of a promising immunotherapy in the treatment of CRCs (Figure 1).
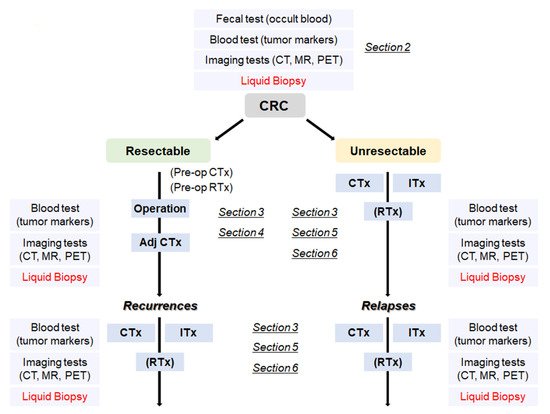
Figure 1. A flowchart from the diagnosis of CRC to treatment according to the disease progression. CRC: colorectal cancer, Adj CTx: adjuvant chemotherapy, CTx: chemotherapy, RTx: radiotherapy, Itx: immunotherapy.
2. Cancer Screening Using Liquid Biopsy
CRC is often diagnosed at a late stage owing to the lack of specific symptoms in early stages [1]. It is clinically important to develop an easy, cheap, and sensitive cancer screening method that detects cancer or precancerous lesions before clinical symptoms arise. If we can, we are able to begin treatment at earlier stages and increase probability of cure of the disease.
Colonoscopy screening remains the gold standard for early-stage diagnosis of CRC and has led to the reduction of CRC-related mortality [2][3][2,3]. Although colonoscopy is certainly effective, it is an invasive and relatively expensive procedure to screen CRC. Recent advancement of noninvasive screening approaches that were approved by the US Food and Drug Administration (FDA) includes stool-based tests and multitarget stool DNA tests (e.g., Cologuard) summarized in Table 1.
Table 1. Up-to-date stool- and blood-based screening method.
| Screening Method | Type of Sample | Description | Overall Performance | ||
|---|---|---|---|---|---|
| High sensitivity Guaiac-based fecal occult blood test (gFOBT) -FDA approved |
Stool |
| In randomized controlled trials, screening with FOBT reduced CRC mortality rates by 15% to 33% [4][5][6 |
.
On the other hand, Guardant Health has initiated the ECLIPSE study (https://clinicaltrials.gov/ct2/show/NCT04136002, accessed on 26 April 2021) for early detection of CRC with the LUNAR-2 blood test. The LUNAR-2 assay could detect somatic variants, methylation alterations, and other epigenomic changes and reported a high sensitivity in detecting CRC. This assay will be further tested on approximately 10,000 individuals aged 45–84 who are at average risk for CRC.
GRAIL started the Circulating Cell-free Genome Atlas Study (CCGA) as a discovery study and found that whole-genome bisulfite sequencing (WGBS) interrogating genome-wide methylation patterns outperformed whole-genome sequencing (WGS) and targeted sequencing approaches interrogating copy-number variants (CNVs) and single-nucleotide variants (SNVs)/small insertions and deletions, respectively [20][21][20,21]. GRAIL established a high-specificity (low false positive rate) targeted bisulfite sequencing, which focused on more than 100,000 methylation sites in our genome and assessed methylation patterns to evaluate the presence or absence of cancer with machine learning. The results of CCGA and the STRIVE study reported a sensitivity of 67.3% for 12 cancer types at stages I to III with an accuracy of 93% to predict tissue of origin [14].
Cristiano et al. [15] developed a method called DELFI (DNA evaluation of fragments for early interception) for early cancer detection. This method utilized the differences of genome-wide cfDNA fragmentation profiles as well as machine learning to distinguish cancer patients from healthy individuals. DELFI detected 152 of 208 patients with eight cancer types including breast, CRC, lung cancer, ovarian cancer, pancreatic cancer, gastric cancer, and cholangiocarcinoma. The overall sensitivity and specificity were 73% and 98%, respectively and 81% and 95% for CRC, respectively. Furthermore, among the 126 patients who were evaluated by both targeted sequencing and DELFI, the sensitivity of DELFI alone was 66% (83 of the 126 patients), but when combining both tests, the sensitivity improved to 82% (103 of the 126 cases) [15].
3. Genomic Analysis for Selection of Molecular-Targeted Drugs
In cases of systemic/distant recurrences after the curative resection of primary CRCs or in those with surgically unresectable stage IV CRCs, intensive systemic chemotherapy is provided to halt the progression of the disease. However, it is difficult to completely eradicate cancer cells using the current regimens of systemic chemotherapy, so novel therapies based on the genomic profiles of the tumors of individual patients should be developed. Some of targeting genetic mutations include KRAS, BRAF, HER2, and microsatellite instability (MSI), which are leveraged in the current clinical setting [22] (Table 2).
Table 2. Genomic biomarkers in CRCs.
| Gene | Biomarkers | Frequencies (%) | Anticancer Agents | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KRAS | Wild type | 60 [23] | ] Specificity: 86.7% to 97.7%; Sensitivity: 33.3% to 79.4% [7][8] | In randomized controlled trials, screening with FOBT reduced CRC mortality rates by 15% to 33% [4,5,6] Specificity: 86.7% to 97.7%; Sensitivity: 33.3% to 79.4% [7,8] |
||||||||
| Cetuximab | Panitumumab | Fecal Immunochemical test (FIT) -FDA approved |
Stool | |||||||||
| KRAS |
|
G12C | 8 [23] | Randomized control: On-going Specificity: 81% to 96%; Sensitivity: 65% to 95% [9][10] | Randomized control: On-going Specificity: 81% to 96%; Sensitivity: 65% to 95% [9,10] |
|||||||
| Sotorasib (AMG510) | Adagrasib (MRTX849) | Multitarget stool DNA test (Cologuard) -FDA approved |
Stool |
| Specificity: 86.6%; Sensitivity: 92.3% [11] |
|||||||
| Epi proColon | ||||||||||||
| BRAF | V600E | 10 [24] | Vemurafenib Dabrafenib Ecorafenib |
-FDA approved |
Blood | |||||||
| HER2 | Amplification |
| 2–3 [25][26] | 2–3 [25,26] | Specificity: 80 to 82%; Sensitivity: 68 to 72% [12] |
|||||||
| Pertuzumab | Trastuzumab | Lapatinib |
CancerSEEK | Blood | ||||||||
| MLH1 | MSH2 | MSH6 | PMS2 |
| MSI-H | 10–15 [27][28] | 10–15 [27,28] | All 8 cancer types including CRC Specificity > 99% Sensitivity: 33% to 70% [13] |
||||
| Nivolumab | GRAIL (CCGA and STRIVE) | Blood |
| All 12 cancer types including CRC Specificity: 98.3% to 99.8% Stage I–III sensitivity: 60.7% to 73.3% [14] |
||||||||
| DELFI | Blood |
| Specificity: 95%; Sensitivity: 81% [15] |
A follow-up diagnostic colonoscopy is performed if abnormalities are indicated by these noninvasive tests. Even though the existing noninvasive stool-based tests for colon cancer have shown high sensitivity and specificity, adherence remains low. An observational study from Germany indicated the improved compliance to CRC screening using these tests. In this study, 97% of the subjects who had refused colonoscopy accepted an alternative noninvasive method; 83% of them chose the blood test, only 15% chose the stool test, and the remaining people refused to receive any screenings [16].
To improve the low adherence of colonoscopy and stool-based tests, liquid biopsy approaches, which have progressed substantially, are likely to be suitable to apply for the screening of CRC using blood samples. In 2016, the FDA approved the first blood-based screening test, Epi proColon, that possibly detects the promoter methylation status of the septin 9 (SEPT9) gene in cell-free DNAs (cfDNAs) for colon cancer using qPCR. Methylated SEPT9 level is increased in CRC and thus serves as a differential biomarker for early detection of CRC. The Epi proColon assay showed sensitivity and specificity values ranging from 68 to 72% and 80 to 82%, respectively (Table 1) [17][18][17,18]. Importantly, results from the multicenter randomized ADMIT trial indicated that adherence of Epi proColon blood-based screening was 99.5% compared with 88.1% for the FIT stool-based test, demonstrating a preferential acceptance of the blood test [12].
In addition to methylation signatures, Cohen and colleagues reported a multianalyte detection systems (CancerSEEK) by combining the detection of specific mutations of circulating tumor DNA (ctDNA) with conventional biomarkers for the detection of eight common surgically resectable cancer types. The CancerSEEK approach detected cancer with a sensitivity range from 69% to 98% and a specificity of 99% [13]. Specifically, 65% (252/388) of stage I–III resectable CRC were positive with CancerSEEK [13]. A subsequent prospective interventional study, DETECT-A (Detecting cancers Earlier Through Elective mutation-based blood Collection and Testing) evaluated 10,006 women with no prior history of cancer and followed-up for 12 months with the combination of the CancerSEEK study and imaging. Among those participants, 127 of 134 had a positive blood test underwent PET–CT imaging examination to evaluate the presence or absence of as well as the location of cancer. A total of 26 women were diagnosed to have a cancer, and 65% of them were found to be at a localized stage, potentially amenable to surgical resection [19]
| Pembrolizumab |
| Ipilimumab |
As for application of the KRAS mutation status to select chemotherapy regimens, phase III clinical trials such as CRYATAL, OPUS, CO.17, and FIRE-3 have shown that the benefit of adding cetuximab (anti-epidermal growth factor (EGFR) antibody) to FOLFOX or FOLFIRI was confined to patients with CRCs not having KRAS mutations [29][30][31][32][33][34][35][29,30,31,32,33,34,35]. With respect to KRAS-mutant tumors, the complexity of the signaling network of the KRAS-mutant alleles has made it difficult to develop molecularly-targeted therapies against KRAS mutations. Mutant KRAS protein has thus been regarded as an undruggable target, so most therapeutic strategies have been designed to inhibit downstream effector pathways such as the ERK/MAPK cascade. However, the clinical efficacy of targeting downstream effectors has been marginal [36]. Two covalently-binding inhibitors, AMG510 (Sotorasib) and MRTX849 (Adagrasib), which specifically target the KRAS G12C mutation, have recently been developed [37][38][39][40][41][42][37,38,39,40,41,42], and their encouraging efficacy in solid tumors harboring the KRAS G12C mutation including non-small cell lung cancers (NSCLCs) and CRCs has been demonstrated in several clinical trials [43][44][45][46][47][43,44,45,46,47]. However, most NSCLC patients with the KRAS G12C mutant showed a favorable response to selective KRAS G12C inhibition, while CRC patients harboring the same mutation rarely revealed clinical benefits. This drug resistance is speculated to result from a possible mechanism where a novel mutation can appear [48][49][48,49] and/or the feedback reactivation of the RAS pathway following KRAS G12C inhibition may occur. To overcome the acquired resistance by the adaptive RAS pathway feedback reactivation in CRCs, combinatorial targeting of EGFR and KRAS G12C or, theoretically, concomitant inhibition of SHP2 and KRAS G12C is expected as a promising treatment strategy, since SHP2 mediates signaling from multiple receptor tyrosine kinases to RAS, and its inhibition can more comprehensively hamper the feedback reactivation [50][51][50,51].
Regarding the treatment for CRCs with the BRAF V600E mutation, the administration of a BRAF inhibitor (vemurafenib) alone showed only limited clinical efficacy compared to the favorable responses observed in melanoma patients [52][53][52,53]. As in the case of KRAS G12C inhibition, adaptive feedback reactivation of the RAS-signaling pathway is considered to be a major mechanism of therapeutic resistance or poor response. Specifically, BRAF inhibition in cancers with the BRAF V600E mutation led to loss of negative feedback signals through the MAPK pathway in CRCs, resulting in receptor tyrosine kinase-mediated reactivation of MAPK signaling by wild-type RAS and RAF [54][55][56][57][58][54,55,56,57,58]. The concomitant administration of dabrafenib and trametinib therefore has a substantial impact on clinical efficacy in a subset of patients with BRAF-V600E CRCs [59]. Furthermore, combined BRAF + EGFR + MEK inhibitions are tolerable and result in favorable clinical responses and a significantly longer overall survival compared to standard therapy in patients with BRAF-V600E CRCs [60][61][60,61].
Regarding HER2-positive CRCs, HER2-inhibiting antibodies and small molecules can suppress the activity of HER2-amplification or mutations [62]. According to several clinical trials including HERACLES, MyPathway, and DESTINY-CRC01, HER2-targeted therapies including anti-HER2 antibody conjugated with or without cytotoxic drugs showed promising and long-lasting outcome in HER2-positive CRCs that had been refractory to standard treatment [63][64][65][66][67][63,64,65,66,67]. Although HER2 amplification is identified in only 2–3% of CRCs, these results could provide hope to a substantial number of CRC patients who have experienced progression of the disease with the standardized guidelines.
In MSI-high CRCs due to mismatch-repair deficiency, immune checkpoint inhibitors such as pembrolizumab and nivolumab have shown a significant clinical benefit in clinical trials including CheckMate-142 and KEYNOTE-164 [68][69][70][71][72][73][68,69,70,71,72,73]. Furthermore, pembrolizumab monotherapy has led to clinically meaningful improvements in health-related quality of life compared with chemotherapy in MSI-high CRC patients (KEYNOTE-177) [74][75][74,75]. The administration of immune checkpoint inhibitors is thus regarded as a first-line treatment option for this population [76]. However, acquired resistance to anti-PD-1 immunotherapy was reported in a subset of cases where the expression of MHC and/or B2M was reduced or either of these genes were lost in tumor cells, leading to impaired antigen presentation and resulting in immune evasion [77][78][79][77,78,79]. Hence, therapies targeting CTLA4 or PD-1 still have some limitations in treatment efficacy. It is also an intriguing approach to upregulate MHC-I expression to enhance sensitivity to immunotherapy [80].
NGS-based targeted-gene panel tests have recently been used in a clinical setting to identify patients with actionable genetic alterations for enrollment in genotype-matched clinical trials. According to the mutational landscape of metastatic cancers of more than 10,000 patients with clinical sequencing using a comprehensive assay MSK-IMPACT, one or more potentially actionable genetic alterations was detected in 36.7% of the patients, and 11% could be enrolled to genome-guided clinical trials [81]. In the literature covering genomic testing of advanced cancers, a small proportion of patients (4–31%) had a chance to receive genetic-alteration-matched therapy [82][83][84][85][86][87][88][89][90][91][92][93][94][82,83,84,85,86,87,88,89,90,91,92,93,94]. Furthermore, an observational study involving more than 1000 patients showed that overall response rates, time-to-treatment failure, and overall survival were higher with matched targeted therapy than those observed without matching, suggesting that identifying specific genetic alterations and choosing therapy based on these alterations are associated with a better prognosis than standard systemic therapy [83]. This finding is consistent with a recent meta-analysis of phase I trials that showed a higher overall response rate (30.6% vs. 4.9%, p < 0.001) and median progression-free survival (5.7 months vs. 2.95 months, p < 0.001) for genotype-matched trials, compared with non-selected therapies [95].
With the advent of WGS and whole-exome sequencing (WES), we can share more comprehensive information on the genomic alterations of individual tumors than with targeted gene-panel sequencing [22]. A recent WGS analysis comprising 2520 samples in 22 types of metastatic tumors showed that 62% of these tumors harbored at least one actionable mutation [96]. With the advances in sequencing technologies, more actionable biomarkers and/or oncogenic mutations have been detected in individual cancers. While it might suggest that a high proportion of actionable alterations are detectable in cancer patients by WGS and WES, the clinical benefits for cancer patients are still very limited. The limited contribution of gene panel tests is attributable to a variety of reasons ranging from patient-dependent factors such as health deterioration in those with an advanced cancer and patient preferences to physician-dependent factors including strict inclusion criteria of clinical trials, the cost of off-label use of drugs, and limited supply of molecular-targeted drugs [84][85][88][84,85,88]. Since genomic profiles can alter clinical management in diverse scenarios, the combination of comprehensive molecular testing and better access to genome-guided trials can improve rates of clinical trial enrollment, thereby enabling precision cancer medicine on a large scale. To expand opportunities for genome-matched therapies, further development of novel molecular-targeted drugs and other treatment options is urgently required.