3.1. N6-Methyl-Adenosine (m6A)
m6A was first reported in the 1970s in mammalian RNAs
[49][50][51][61,62,63]. A full comprehension of the role of this modification took several decades. In 1997, the protein Methyltransferase-like (METTL) 3 was identified as the first “writer” of m6A in mammalian cells
[20][23]. Further investigations have shown that m6A is installed by a nuclear complex comprised of METTL3, METTL14 and WT1-Associated Protein (WTAP)
[19][22]. Further components of this complex include KIAA1429, RNA Binding Motif Protein 15 (RBM15) and Zinc Finger CCCH-Type Containing 13 (ZC3H13)
[52][53][54][64,65,66].
Several members belonging to the YTH (YT521-B homology) family, such as human YT521-B (also known as YTHDC1), YTHDC2, YTHDF1, YTHDF2 and YTHDF3, have been identified as m6A-binding or “reader” proteins
[15][22][18,25]. The members belonging to the DF family likely confine m6A-modified RNAs in specific cytoplasmic liquid–liquid phase separation compartments
[55][67].
Several other “reader” proteins have been shown to bind m6A-modified RNAs thanks to a so-called “m6A switch”
[56][68]. Indeed, m6A installation may trigger a conformational switch that allows the binding of these “reader” proteins, which, in fact, do not directly bind to the m6A residue itself
[10][13]. This mechanism is exploited by several members of the hnRNP (heterogeneous nuclear ribonucleoprotein) family. Finally, insulin-like growth factor 2 mRNA-binding proteins (IGF2BP) were also reported to bind m6A-modified RNAs, promoting their stability
[57][69].
Although two enzymes able to “erase” m6A from mammalian RNAs have been reported, i.e., FTO Alpha-Ketoglutarate-Dependent Dioxygenase (FTO) and AlkB Homolog 5, RNA Demethylase (ALKBH5)
[26][27][29,30], the specificity and the relevance of these enzymes in physiological conditions are still a matter of debate
[58][70].
About 0.1–0.4% of all adenosines in global cellular RNAs are modified as m6A, and this modification accounts for ~50% of all methylated ribonucleotides
[49][61]. m6A was found in all classes of cellular RNAs: mRNAs (in particular, in long internal exons, locations upstream of stop codons and the 3′-UTR regions)
[22][59][60][25,71,72]; ribosomal RNAs; transfer RNAs and various non-coding RNAs
[61][62][63][73,74,75].
In cancer, the relevancy of m6A in miRNA maturation was first unveiled for miR-126 in hepatocellular carcinoma (HCC) [64][82].
From that moment on, increasing evidence has disclosed the relevance of the m6A modification of miRNA in cancer progression. Most of the literature confirms that m6A mainly promotes pri-miRNA processing and that the deregulation of the enzymes involved in writing or reading m6A is correlated with tumor onset. Notably, alteration of the m6A deposition on miRNAs is not only a common feature of different tumors but also participates in tumorigenesis processes (
Figure 1 and
Table 1).
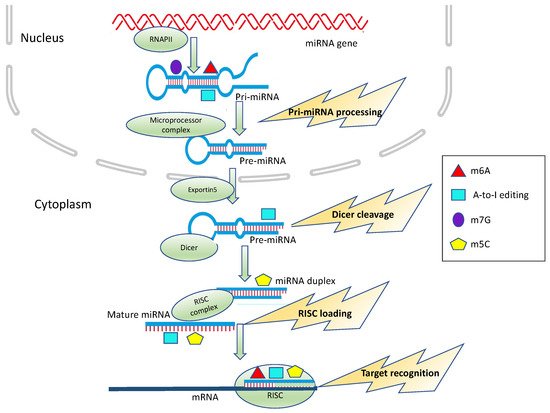
Figure 1. Epitranscriptomic modification impacts on miRNA processing and activity. m6A, A-to-I editing, m5C and m7G can affect different steps of miRNA biogenesis, including Microprocessor cleavage, Dicer cleavage and RISC loading or alter target recognition and binding.
Table 1. Effects of m6A modification of miRNAs in cancer.
|
| Cancer Type |
|
| m6A-Modified |
| miRNA(s) |
|
| Increase/ |
| Decrease 1 |
|
| Effects on miRNA Processing/Function |
|
| Effects on Tumor Progression |
|
| Reference |
|
|
| Colorectal Cancer |
|
| miR-1246 |
|
| ↑ |
|
| processing |
|
| Up-regulation of mature of miR-1246 results in the reduction of SPRED2, thus activating the RAF/MEK/ERK pathway |
|
|
| Binding to targets |
|
| Unedited miR-376a-5p promotes aggressive glioma growth, by its ability to target RAP2A and concomitant inability to target AMFR |
|
[65]
|
[85]
|
|
| [ | 106 | ] |
|
[125]
|
| miR-375 |
|
|
|
| miR-221/222 |
| miR-21↓ |
|
| processing |
|
|
| ↓ |
| Down-regulation of mature miR-375 increases the expression of its targets YAP1 and SP1 thus increasing proliferation, and migration and invasion |
|
|
| processing |
|
| Up-regulation of mature miR-221/222 and miR-21 results in the repression of its targets p27Kip1 and PDCD4, thus increasing proliferation and migration of glioblastoma |
|
[66]
|
[83]
|
|
| [ | 101 | ] |
|
[118]
|
| miR-483, |
| miR-676 |
| miR-877 |
|
| n.d. |
|
| processing |
|
| miR-483, miR-676 and miR-877 modulate mitochondrial metabolism by targeting electron transport chain genes |
|
| miR-589-3p |
|
|
| ↓ |
|
| Binding to targets |
|
[ |
| Editing within miR-589–3p retargets the miRNA from the protocadherin PCDH9 to the metalloprotease ADAM12, which is involved in glioblastoma cell invasion. |
| 67]
|
[90]
|
|
| [ | 107 | ] |
|
[129]
|
| miR-17-5p |
| let-7a-5p |
|
| ↑ |
|
| Binding to targets |
|
| n.d. |
|
[68]
|
[91]
|
|
| Melanoma |
|
| miR-455-5p |
|
| ↓ |
|
| Binding to targets |
|
| Unedited miR-455-5p but not the edited form targets the tumor suppressor gene CPEB1, thus promoting tumor growth and metastasis |
|
[108]
|
[127]
|
| Pancreatic cancer |
|
|
| miR-378a-3p |
|
| miR-25-3p |
|
| ↓ |
|
| ↑ |
|
|
| Binding to targets |
| processing |
|
| Up-regulation of mature miR-25-3p results in the reduction of PHLPP2, leading to AKT activation. |
|
| Edited miR-378a-3p but not the unedited form specifically targets the PARVA oncogene, thus preventing the progression of melanoma towards the malignant phenotype |
|
[69]
|
|
[[89] |
|
109 | ] |
|
| [128]
|
| miR-17-5p |
| let-7a-5p |
|
| ↑ |
|
|
|
| Chordoma |
|
| miR-10a |
| miR-125a |
| Binding to targets |
|
| ↑ |
| n.d. |
|
| processing |
|
[68]
|
[91]
|
|
| Down-regulation of miR-10a and miR-125a expression and upregulates expression of their target genes |
|
| [ | 102]
|
[119]
|
| Hepatocellular Carcinoma |
|
| miR-126 |
|
| ↓ |
|
| processing |
|
|
| Chronic myeloid leukemia |
|
| let-7 |
|
| ↑ |
| Down-regulation of mature miR-126 which acts as a tumor suppressor |
|
|
| processing |
|
[64 |
| Down-regulation of mature let-7 results in increased LIN28B expression and enhanced self-renewal |
| ]
|
[82]
|
|
| [ | 103 | ] |
|
[120]
|
| Bladder cancer |
|
| miR-221/222 |
|
|
|
| Thyroid cancer |
| ↑ |
|
|
| miR-200b |
| processing |
|
| Up-regulation of mature miR-221/222 results in the reduction of PTEN, leading to proliferation |
|
| ↑ |
|
[70]
|
[ |
| Binding to targets |
84]
|
|
| Edited miR-200b has weakened activity against its target gene ZEB1, an epithelial–mesenchymal transition (EMT) marker |
|
| [ | 105]
|
[131]
|
| Gallbladder cancer |
|
| miRNA-92 |
|
| ↑ |
|
| processing |
|
| Up-regulation of mature miRNA-92 results in the reduction of PTEN, thus activating PI3K/AKT signaling |
|
[71]
|
[88]
|
|
| Ovarian cancer |
|
| miR-126 |
|
| ↑ |
|
| processing |
|
| Up-regulation of mature miR-126-5p results in the reduction of PTEN, thus activating the PI3K/Akt/mTOR pathway |
|
[72]
|
[87]
|
|
| Gastric cancer |
|
| miR-17-5p |
| let-7a-5p |
|
| ↑ |
|
| Binding to targets |
|
| n.d. |
|
[68]
|
[91]
|
|
| Lung cancer (brain metastasis) |
|
| miR-143-3p |
|
| ↑ |
|
| processing |
|
| Up-regulation of mature miR-143-3p promotes the metastatic potential of lung cancer via regulation of angiogenesis and microtubules through VASH1 |
|
[73]
|
[86]
|
1 increase (↑) or decrease (↓) of the epitranscriptomic modification (n.d., not detected; SPRED2, Sprouty Related EVH1 Domain Containing 2; YAP1, yes-associated protein 1; SP1, Sp1 Transcription Factor; PHLPP2, PH Domain And Leucine Rich Repeat Protein; PTEN, Phosphatase 2 Phosphatase And Tensin Homolog; VASH1, Vasohibin 1.
3.2. A-to-I Editing
A-to-I editing is catalyzed by enzymes highly conserved in vertebrates, called Adenosine Deaminases Acting on RNA (ADAR)
[74][92]. Mammalian genomes encode for three members of the ADAR family: ADAR1, ADAR2 and ADAR3
[75][93].
ADAR enzymes bind double-stranded (ds) regions of coding and noncoding RNAs
[76][94]; in RNAs forming imperfect dsRNA structures, A-to-I editing involves only one or two adenosines (site selective editing), while, in the case of long perfect dsRNA regions, the random modification of several A residues is observed (hyper-editing)
[77][78][79][95,96,97].
Inosine is recognized by the cellular machinery as guanosine, causing a change in the RNA sequence. As a consequence, depending on the modification site, this type of RNA editing can influence the RNA stability
[80][81][82][98,99,100], splicing
[83][84][85][101,102,103], localization and translation, as well as redefine its interactions with specific factors
[86][87][104,105]. In mRNAs, the modification of A-to-I can lead to a codon change, thus affecting the primary structure of the encoded protein
[88][89][106,107].
A-to-I editing mainly targets noncoding regions of RNA, such as introns and UTRs, containing repetitive Alu elements and Long Interspersed Elements (LINEs) that fold into dsRNA structures recognized by ADARs
[90][108].
In most types of cancer, the activity of ADAR enzymes is significantly decreased, as witnessed by the extensive hypoediting of Alu RNAs, as well as by the reduced expression of ADAR enzymes
[91][109].
The first evidence of the editing of a miRNA was shown in 2004 by Luciano and colleagues
[92][33], who reported A-to-I conversion within the miR-22 precursor in
Homo sapiens and
Mus musculus. Soon after, it was shown that the A-to-I editing of pri-miR-142 prevents processing by DROSHA
[93][110]. ADAR enzymes have a degree of specificity for different miRNA precursors, depending on their secondary structure
[94][111]. The ADAR1 interaction with DICER was associated with enhanced miRNA processing in oral squamous cells carcinoma
[95][112] and in melanoma
[96][113], although, in both cases, the authors did not assess the editing of the miRNA precursors. Furthermore, ADAR editing has been shown to affect the DICER-dependent processing of viral miRNAs
[97][114]. Of note, ADARs can also alter miRNA metabolism independently from their editing activity
[98][99][100][115,116,117].
Several examples showed that the A-to-I editing of miRNA precursors inhibits the biogenesis of mature miRNAs or
alters the selection of miRNA targets (
Figure 1 and
Table 2). The deregulation of ADAR1 and/or ADAR2 in glioblastoma and in chordoma affects the expression levels of miR-21, miR-221 and miR-222
[101][118] and of miR-10a and miR-125a
[102][119], respectively. Furthermore, the impairment of let-7 biogenesis by means of ADAR1-mediated A-to-I editing drives leukemia stem cells renewal
[103][120].
In thyroid cancer, the slight overexpression of ADAR1 corresponds to a higher expression of ZEB1, a master regulator of Epithelial–Mes- enchymal Transition (EMT). It has been demonstrated that editing of the seed sequence of miR-200b by ADAR1 impairs its ability to inhibit ZEB1 expression, favoring the progres- sion of the cancer [104][105][130,131].
Table 2. Effects of A-to-I editing of miRNAs in cancer.
|
| Cancer |
|
| A-to-I-Modified |
| miRNA(s) |
|
| Increase/ |
| Decrease 1 |
|
| Effects on miRNA Processing/Function |
|
| Effects on Tumor Progression |
|
| Reference |
|
|
| Glioma |
|
| mir-376a-5p |
|
| ↓ |
|
|
Lung cancer |
|
|
|
|
miR-381 |
|
|
| ↑ |
|
| n.d. |
|
| Edited miR-381 enhances the growth of non-small-cell lung cancer cells as compared to the unedited form |
|
[110]
|
[134]
|
1 increase (↑) or decrease (↓) of the epitranscriptomic modification (n.d., not detected; RAP2A, Ras-Related Protein Rap-2a; AMFR, Autocrine Motility Factor Receptor; PDCD4, Programmed Cell Death 4; PCDH9, Protocadherin 9; ADAM12, ADAM Metallopeptidase Domain 12; CPEB1, Cytoplasmic Polyadenylation Element-Binding Protein 1; PARVA, Parvin Alpha).
3.3. 5-Methylcytosine (m5C)
m5C is one of the most representative post-transcriptional RNA modifications
[111][140], and it has long been known to be present in all three kingdoms of life
[112][113][141,142].
m5C was originally reported in tRNAs, rRNAs
[50][62] and coding RNAs
[114][143]; later, it was identified in other noncoding RNAs, thanks to technologies such as bisulfite treatment and Next-Generation Sequencing (NGS)
[115][116][117][144,145,146].
The synthesis of m5C is catalyzed by the seven members of the NOL1/NOP2/SUN domain (NSUN) family of methyltransferases
[118][147] or by DNA methyltransferase-2 (DNMT2)
[119][148]. These enzymes are responsibles for the methylation of rRNAs, tRNAs
[120][121][122][123][124][149,150,151,152,153], mRNAs
[125][126][127][154,155,156], lncRNAs
[128][157], vault-RNAs
[129][158], enhancer-RNAs
[116][145], mitochondrial tRNAMet
[130][159] and mitochondrial 12S rRNA
[131][160].
In vitro and in vivo studies have demonstrated that aly/REF nuclear factor (ALYREF) is a putative “reader” of m5C sites on mRNAs and that, following the knockdown of NSUN2, ALYREF loses its RNA-binding ability and is retained in the nucleus, suggesting a role for m5C in mRNA exports from the nucleus
[23][26]. A further m5C “reader” is Y-Box-Binding Protein 1 (YBX1) that recognizes and binds m5C-modified mRNAs and stabilizes their target mRNAs by recruiting ELAV-like Protein 1 (ELAVL1)
[132][133][161,162].
m5C “writers” and “readers” are primarily implicated in fundamental cancer-related processes such as cell differentiation, motility
[134][135][163,164], proliferation
[136][137][165,166], cell cycle progression
[138][167] and senescence
[126][155].
In particular, NSUN2 is aberrantly expressed and plays important roles in the development and pathogenesis of different types of tumors, such as breast, colorectal, lung, skin, ovarian and bladder cancers
[139][168].
The distribution of m5C in small RNAs is poorly understood so far; nevertheless, this modification has been recently highlighted in vault RNAs (vtRNAs)
[129][158], piwi-associated RNAs (piRNAs)
[140][169] and miRNAs
[68][141][142][91,170,171]. m5C deposition regulates the processing of vault ncRNAs into small vault RNAs (svRNAs)
[129][143][158,172].
m5C has been only recently characterized in miRNAs. Interestingly, methylation, but not an abundance of miR-200c-3p and miR-21-3p, was increased in pancreatic and colorectal cancer tissues, as well as in serum samples from pancreatic and colorectal cancer patients
[68][91] (
Figure 1 and
Table 3).
Recently, we described that m5C is widely spread in human miRNAs in various sequence contexts by taking advantage of a novel NGS analysis of bisulfite-treated small RNAs (BS-miRNA-seq) [142][171]. In this context, not only the presence of m5C but, also, of hm5C on several miRNAs in human cancer cell lines was unraveled.
Table 3. Effects of m5C and m7G modifications of miRNAs in cancer.
|
| Cancer |
|
| Modified |
| miRNA(s) |
|
| Increase/ |
| Decrease 1 |
|
| Effects on miRNA Processing/Function |
|
| Effects on Tumor Progression |
|
| Reference |
|
|
| Glioma |
|
| miRNA-181a-5p |
| (m5C) |
|
| ↑ |
|
| Binding to targets |
|
| Cytosine-methylated miRNA-181a-5p loses its ability to target the mRNA of the pro-apoptotic protein BIM |
|
[140]
|
[169]
|
|
| Colorectal cancer; gastric cancer; pancreatic cancer |
|
| miR-200c-3p |
| miR-21-3p |
| (m5C) |
|
| ↑ |
|
| Binding to targets |
|
| n.d. |
|
[71]
|
[88]
|
|
| Lung cancer |
|
| let-7 family |
| (m7G) |
|
| n.d. |
|
| processing |
|
| m7G methylation within miRNAs regulates cell migration |
|
[144]
|
[173]
|
|
| Colon cancer |
|
| let-7e |
| (m7G) |
|
| ↓ |
|
| processing |
|
| Down-regulation of mature let-7e results in the activation of its targets HMGA2 thus stimulating colon cancer cell viability and mobility |
|
[145]
|
[174]
|
1 Increase (↑) or decrease (↓) of epitranscriptomic modifications (n.d., not detected; HMGA2 High Mobility Group AT-hook 2).
3.4. N7-Methylguanosine (m7G)
m7G is a positively charged modification installed cotranscriptionally at the 5’ Caps of eukaryotic mRNAs
[146][182]. This modification protects and stabilizes transcripts from exonucleolytic degradation
[147][183] and influences all the events responsible for the processing of the mRNA molecules, from transcript elongation to translation
[148][149][184,185].
Notably, the presence of internal m7G sites was found not only in tRNA and rRNA molecules
[150][151][152][186,187,188] but also in mammalian mRNAs
[152][188]. Internal m7G could affect mRNA translation, and this modification typically occurs near the start and stop codons in a GA-enriched motif
[153][189].
The enzyme responsible for this internal m7G modification is METTL1, which cooperates with the cofactor WD Repeat Domain 4 (WDR4)
[153][154][189,190]. Interestingly, METTL1 has been linked to tumor vascular invasion and poor prognosis in hepatocellular carcinoma
[144][155][173,191].
Recently, by high-throughput screening, several miRNAs were identified as harboring internal m7G sites
[156][192]. In particular, METTL1-dependent m7G was discovered in a subset of tumor-suppressor miRNAs involved in the inhibition of cell migration, including the let-7 family. METTL1-mediated m7G occurs on pri-miRNA within G-rich regions that display the propensity to form G-quadruplexes, i.e., structures known to be inhibitory to miRNA processing
[145][157][158][174,193,194] (
Figure 1 and
Table 3).
Indeed, m7G in the let-7 family affects G-quadruplex formations, thus facilitating the formation of a canonical stem-loop structure and miRNA processing [156][192]. In line with this study, Liu and colleagues showed that, in colon cancer, the downregulation of METTL1 leads to a decrease in the let-7e levels. The alteration of let-7e expression affects its downstream target High Mobility Group AT-hook 2 (HMGA2), thus promoting cell proliferation, invasion and EMT [159][195].