Regulatory SNPs are genetic variants that associated with various human traits and diseases map to a noncoding part of the genome and are enriched in its regulatory compartment, suggesting that many causal variants may affect gene expression. The leading mechanism of action of these SNPs consists in the alterations in the transcription factor binding via creation or disruption of transcription factor binding sites (TFBSs) or some change in the affinity of these regulatory proteins to their cognate sites.
1. Introduction
A central goal of human genetics is to understand how genetic variation leads to phenotypic differences and complex diseases. Recently, genome-wide association studies (GWAS) have detected over 70 thousand variants (mainly, single nucleotide polymorphisms, SNPs) associated with various human traits and diseases
[1][2]. The vast majority of the genetic variants identified from GWAS map to the noncoding part of the genome and are enriched in regulatory regions (promoters, enhancers, etc.), suggesting that many causal variants may affect gene expression
[3][4][5][6].
As is known, the regulatory regions of the genome represent clusters of the binding sites for sequence-specific transcription factors (TFs). There, the interplay between these TFs and their binding sites (
cis-regulatory elements) as well as the interaction of TFs with one another and the coactivator and chromatin remodeling complexes orchestrate the dynamic and diverse genetic programs, thereby determining the tissue-specific gene expression, spatiotemporal specificity of gene activities during development, and the ability of genes to respond to different external signals
[7][8][9][10][11][12]. Thus, thanks to the binding to their specific sites on DNA (transcription factor binding sites, TFBSs), TFs directly interpret the regulatory part of the genome, performing the first step in deciphering the DNA sequence
[13][14][15]. Consequently, regulatory SNPs (rSNPs), that is, genetic variation within TFBSs that alters expression, play a central role in the phenotypic variation in complex traits, including the risk of developing a disease.
Starting from the 1990s, numerous studies have been performed focusing on the noncoding SNPs that perturb the TF binding and are associated with various pathologies. As has been shown, risk alleles can (i) destroy a binding site for a TF
[16][17][18][19]; (ii) create a binding site for a TF
[20][21][22]; or alter the binding affinities towards an increase
[23][24][25] or a decrease
[25][26][27][28]. In addition, several cases have been observed when a damage/destruction of a binding site for a TF leads to a concurrent formation of another/other TFBS(s)
[19][29][30].
The advent of the NGS technologies gave a strong impetus to the development of functional genomics and application of its methods to the genome-wide search for rSNPs. Currently, various methods of functional genomics are used for both mass interpretation of GWAS data and independent genome-wide identification of regulatory variants. So far, expression quantitative trait locus (eQTL) mapping and identification of allele-specific expression (ASE) events utilizing analysis of RNA-seq data (actually, the largest available genome-wide dataset) are the major relevant methods. The search for allele-specific binding (ASB) events in the data of DNase-seq, ChIP-seq, ATAC-seq (assay for transposase-accessible chromatin with high-throughput sequencing), and so on becomes ever more important. In addition, the approaches not directly associated with obtaining genome-wide data are actively used, including massively parallel reporter assay (MPRA), SNPs-seq, and SNPs-SELEX.
2. rSNPs on a Genome-Wide Scale
Genome-wide approaches to the search for rSNPs fall into two large groups. The first group comprises GWAS mass data analysis utilizing manifold methods of functional genomics, while the second group uses the same methods but independently without any prior knowledge about trait associations (Figure 1, Table 1). The latter group includes eQTL analysis, identification of allele-specific events, and some other genome-wide approaches. As for the rSNPs discovered by the approaches of the second group, it is necessary to additionally determine their association with a certain trait (most frequently, via comparison with GWAS data or by analysis of rSNPs as an eQTL in transcriptome data and reconstruction of the gene networks and molecular pathways).
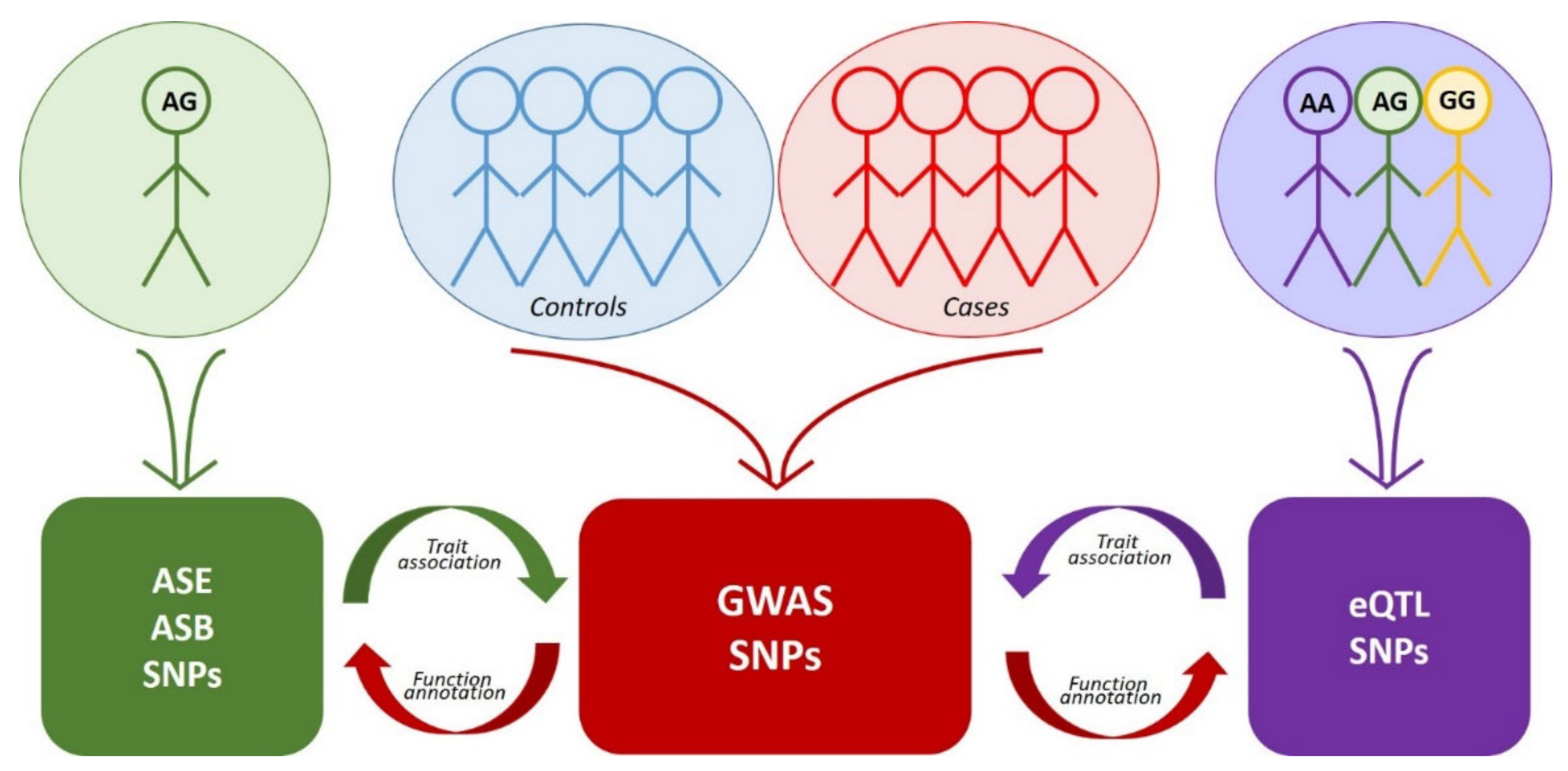 Figure 1.
Figure 1. Interplay between the approaches to the search for functional SNPs. Colored blocks denote arrays of corresponding data. Red arrows indicate functional annotation of GWAS data using eQTL, ASE or ASB analysis. Purple arrow indicates the search for association of eQTL SNPs with traits via comparison with GWAS data. Green arrow—the same for SNPs detected by ASE or ASB analysis.
Table 1. Main features of the most widespread approaches to the search for functional SNPs.
| |
Approach |
GWAS |
eQTL Analysis |
ASE |
ASB |
| 1 |
Initial association
with trait |
+ |
− |
− |
− |
| 2 |
Initial association
with function |
− |
+ |
+ |
+ |
| 3 |
Causal or in LD |
Both |
+ |
++ |
+++ |
| 4 |
Number of participants |
Tens and hundreds of thousands
(large cohorts) |
Hundreds
(modestly sized cohorts) |
Few |
Few |
3. Conclusions
Gene expression programs underlying development, differentiation, and environmental responses are guided by the regulatory DNA portion of the metazoan genomes. The corresponding information encoded in regulatory DNA is actuated via the combinatorial binding of sequence-specific TFs to regulatory regions (cis-regulatory modules, CRMs). CRMs switch on promoters and enhancers and are actually the assemblies of TFBSs arranged to provide particular functions
[10][11][14][31][32][33].
The SNPs located in transcriptional regulatory regions can alter gene expression, which may be either adaptive or lead to a disease. The main mechanism underlying the action of these SNPs consists in changes of TF binding, which comprises creation or disruption of TFBSs (cis-regulatory elements) or alteration of the affinity of TFs for their cognate sites
[34][35][36][37]. Although many SNPs with such properties have been so far discovered, their mass search in genomes remains challenging. This is mainly associated with the tissue, developmental, and environmental specificities in the effects of rSNPs, which is a direct consequence of the corresponding specificities of the harboring cis-regulatory elements
[34][38][39]. Thus, myriads of omics experiments are necessary for this purpose; however, this is still too expensive and time-consuming. The computer methods for recognition of TFBSs in DNA sequences are free of this disadvantage but yet ineffective in detection of both TFBSs and the SNPs changing these sites without the cooperation with omics experiments. The objective reasons here are a high degeneracy of the regulatory DNA code
[15][40][41][42]; high importance of low-affinity sites in gene regulation
[43]; the presence of structural variants of the binding sites for the same TF
[44][45][46][47]; and even nonconsensus TFBSs
[48][49]. All these facts considerably decrease the efficacy of the available methods for TFBS recognition, most of which are based on the PWM model, which oversimplifies the mechanisms underlying TF–DNA interaction
[50][51][52][53]. Development of new generation bioinformatics approaches relying on machine learning and neural networks raises the hope for more efficient and accurate recognition of both the TFBSs and rSNPs in the genomes
[54][55][56][57][58][59].
Thus, despite the achieved progress, we are still at the beginning of the way to comprehensive annotation of the genome regulatory portion, full cataloging of rSNPs, and clarification of their association with molecular phenotypes and, eventually, with various complex traits, including diseases. The further advance requires improving the efficiency of the existing experimental and bioinformatics methods of systems biology and advent of the new relevant approaches.
 +1 point
+1 point





