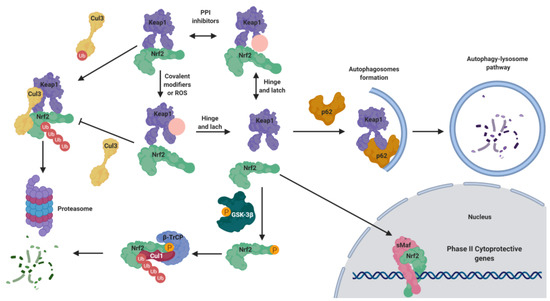
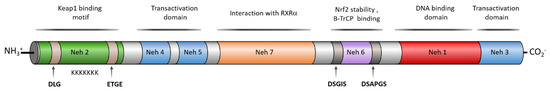
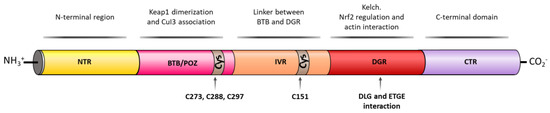
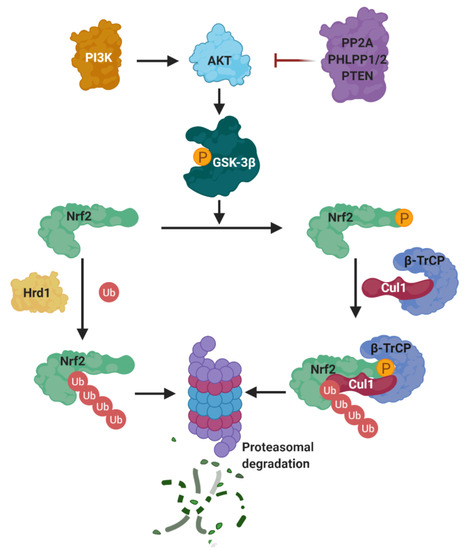
In the past years, many natural and synthetic compounds have been described to potentially activate the NRF2 pathway through this mechanism, and these are summarized below.
Natural and Semisynthetic NRF2 Activators and Their Analogues
Caffeic acid (
2, ) is a polyphenol present in coffee that bears a Michael acceptor embedded in its structure. Some studies reveal that this moiety is responsible and essential for its NRF2 induction properties while its nucleophilic functional groups (catechol) provide clearance but are not directly involved in NRF2 induction
[52]. Further studies have shown that caffeic acid is also able to decrease KEAP1 expression to activate NRF2, leading to an increase in the expression of HO-1 (heme oxygenase 1) and NQO1 (NAD(P)H:quinone oxidoreductase 1)
[52].
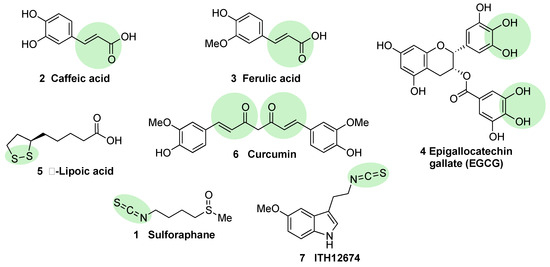
Figure 4. Chemical structures of natural NRF2 activators and their analogues. Caffeic acid (2), ferulic acid (3), and curcumin (6) are considered classical Michael acceptors where a double bound is conjugated with a carbonyl group, creating a potent electrophile. Epigallocatechin gallate (4) is a polyphenolic compound that is able to generate quinone derivatives after oxidation, and the corresponding quinone exerts a potent electrophilic character. Lipoic acid (5) is considered an electrophile that is able to react with KEAP1 Cys residues to form lipoylcysteinyl disulfides, thus modifying their structure. Finally, isothiothianate moieties at sulforaphane (1) and its hybrid melatonin derivative, ITH12674 (7), are responsible for the potent electrophilic character of these compounds. The electrophilic moieties or promoieties of each compound are highlighted in green.
Ferulic acid (
3, ) is widespread in vegetables, and its main structural difference with caffeic acid is one methoxy group on its benzene ring. It has been proved that ferulic acid is able to regulate the NRF2 pathway and counteract trimethyltin (TMT)-induced neuronal damage in the human neuroblastoma cell line SH-SY5Y
[52]. Moreover, it is able to antagonize oxidative stress by ROS scavenging and activating the non-homologous end-joining DNA repair process
[52].
Epigallocatechin gallate (EGCG) (
4, ), a catechin found in green tea
[53], upregulates the NRF2 pathway through electrophilic disruption via its prior auto-oxidation of its catechol moieties to ortho-quinones
[54], as well as via activation of the p38-MAPK and extracellular signal-regulated kinases (ERK)1/2 signaling pathways
[55]. EGCG has shown multiple in vivo and in vitro neuroprotective effects in different models of MS, PD, and traumatic brain injury, all of them associated with an increase in NRF2 induction, antioxidant activity, and a decrease of inflammatory responses
[56][57][58].
Alpha lipoic acid (
5, ) can be found in a wide number of plants including carrots, beets, broccoli, and spinach. The exact mechanism through which it induces NRF2 is not currently known, although in view of its structure, it can form lipoylcysteinyl disulfides with KEAP1, preventing NRF2 degradation
[59][60]. Besides this potential mechanism, it has also been shown that alpha lipoic acid can activate protein kinase C (PKC), which is one of the kinases that is able to activate NRF2 through an alternative pathway
[61]. Several studies on alpha lipoic acid neuroprotective effects have been carried out, although in many of them, it was not investigated whether NRF2 activation took part in these effects. In mice MS models, alpha lipoic acid reduced inflammation
[62]. In addition, in PD models, it decreased ROS, restored ATP levels, preserved dopaminergic neurons, and upregulated mitochondrial formation
[63].
Curcumin (
6, ) is the main curcuminoid present in turmeric acid; it has potent antioxidant and anti-inflammatory properties
[64][65]. It activates NRF2 by electrophilic modification of Cys-151 in KEAP1, modifying also its ROS-scavenging activity
[66]. On the other hand, curcumin is also able to activate NRF2 by repressing KEAP1 expression
[67]. Curcumin showed beneficial neuroprotective activities in intracerebral hemorrhage as well as in traumatic brain injury models
[23][68]. A decreased in proinflammatory gene expression through the prevention of NF-kB activation in microglial cells was also observed upon treatment with curcumin
[69].
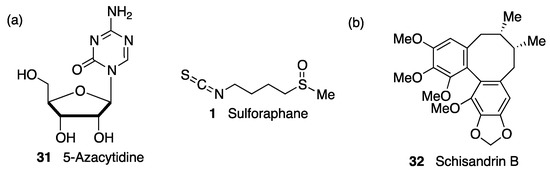
Sulforaphane (
1, ) is an isothiocyanate produced from the enzymatic cleavage of the organosulfur compound glucoraphanin found in cruciferous plants such as broccoli, brussels sprouts, cauliflower, and cabbage
[7]. The catalytic reaction needed for the release of sulforaphane is driven by a β-thioglucosidase present in the gut microbiota
[70]; thus, the levels of released sulforaphane are highly dependent on dietary habits and microbiome composition
[70]. Sulforaphane directly interacts with the Cys151 residue in KEAP1, thereby promoting NRF2 activation
[71][72]. Either in pure form or as sprout extract, sulforaphane has shown a very good safety profile, and more than 30 clinical studies against chronic diseases have been carried out
[73][74]. It has the ability to cross the blood–brain barrier (BBB), and it has shown neuroprotective capacity against Aβ
1-42 peptide in neuronal cells
[75]. In vivo, sulforaphane alleviates cognitive impairment in mouse models of AD
[76]. In PD models, it protected dopaminergic neurons against the parkinsonian toxin 6-hydroxydopamine
[77]. Furthermore, sulforaphane is able to reduce the levels of phosphorylated tau and increase Beclin1 and LC3-II, leading to the conclusion that NRF2 activation may facilitate tau degradation through autophagy
[78]. Nevertheless, sulforaphane is an oily substance with low stability in hydrophilic media, and its pharmacokinetic properties need to be improved
[7]. In this regard, sulforadex (SFX-01), a sulforaphane-β–cyclodextrin complex, has shown an excellent bioavailability and is under clinical trials for the treatment of subarachnoid hemorrhage and metastatic breast cancer
[7]. We will finally mention that sulforaphane has been hybridized with the natural antioxidant melatonin generating ITH12674 (
7, ), which is a compound designed to have a dual drug–prodrug mechanism of action for the treatment of brain ischemia
[79].
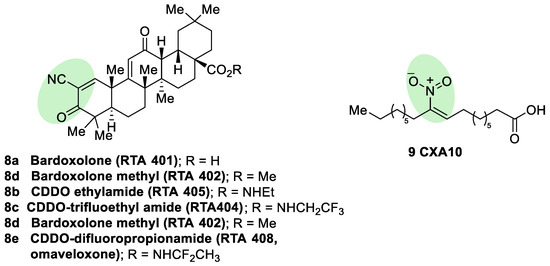
Semisynthetic cyanoenone triterpenoids derived from bardoxolone (RTA401, CDDO,
8a, ), described as antioxidant inflammation modulators (AIMs), exhibit a Michael acceptor moiety and are among the most potent known electrophilic NRF2 activators
[80], which interact with Cys151 in KEAP1 and are under clinical development by Reata Pharmaceuticals and Kyowa Hakko Kirin. Proof-of-concept studies strongly support the use of these triterpenoids for degenerative diseases
[80]. For instance, CDDO-ethyl amide (RTA405,
8b, ) and CDDO-trifluoethyl amide (RTA404,
8c, ) induced a significant reduction of toxicity levels in PD models
[30]. CDDO-methyl ester (CDDO-Me, RTA402,
8d, ) was the first CDDO to reach clinical trials for the treatment of diabetic nephropathy, although it was later withdrawn at phase III (BEACON trial) due to cardiovascular safety issues that are not related with NRF2 induction
[81]. In an effort to improve the safety profile, CDDO-difluoropropionamide (RTA408, omaveloxone,
8e, ) was synthetized and is currently in phase II trial for the treatment of FRDA, ocular inflammation, and pain after ocular surgery
[82].
Figure 5. Chemical structures of some semisynthetic cyanoenone triterpenoids (8) and a nitro fatty acid (9). These compounds are considered electrophilic NRF2 inducers due to the presence of α,β-unsaturations conjugated with carbonyl, cyano, or nitro groups. These electrophilic moieties are highlighted in green.
Nitro fatty acids (NO
2-FAs) are endogenous signaling mediators with anti-inflammatory and antifibrotic activities in preclinical animal models of metabolic and inflammatory disease
[83]. The nitro alkene structure confers electrophilicity to their β-carbon, enhancing the formation of Michael adducts with nucleophiles such as cysteines. It has been proven that NO
2-FAs react with Cys-273 and Cys-288 of KEAP1, activating NRF2
[84]. The reversibility of this reaction
[85] prevents the formation of stable adducts, which could lead to toxicity. Furthermore, compound
9 (10-NO
2-OA, CXA10, ) proved to be safe in humans at an active dose in a phase I safety assay
[86], and it is currently being tested for the treatment of focal segmental glomerulosclerosis.
Synthetic NRF2 Activators
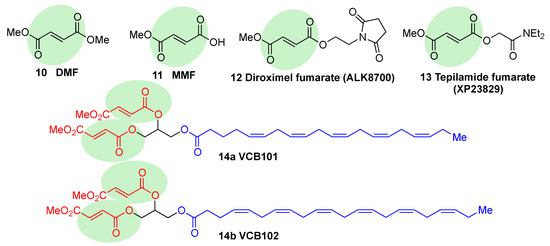
Fumaric acid esters are the group of synthetic NRF2 activators that are receiving the highest amount of attention from the pharmaceutical industry
[18]. Dimethyl fumarate (DMF) (
10, ) is the most clinically successful member of this family. It was firstly approved in 1994 for the treatment of psoriasis
[87], but due to its efficacy in multiple sclerosis mouse models, it was repurposed in 2013 by Biogen under the name Tecfidera for the treatment of relapsing-remitting MS
[88], and it has become one of the most successful new medicines in the past few years
[7]. Due to its thiol-reactive Michael acceptor structure, DMF primarily activates NRF2 via cysteine modification on KEAP1
[89], and it has also been proven to affect NRF2 phosphorylation via the phosphatidylinositol-3-kinase (PI3K) and ERK1/2 pathways
[90].
Figure 6. Chemical structures of fumaric acid ester derivatives. Their electrophilic moieties are highlighted in green.
DMF has shown to be protective in a wide range of neurodegenerative conditions in an AD model
[91] and to prevent hippocampal injury after ischemia and protect BBB integrity in mouse models of stroke
[92]. Furthermore, protection against α−Syn and Aβ toxicity and a reduction in tau hyperphosphorylation
[93][94][95] was also observed.
DMF is mostly converted into monomethyl fumarate (MMF,
11, ) by intestinal esterases. Moreover, it has been shown that MMF reacts with Cys151 in KEAP1, thereby activating NRF2
[96]. Several biopharmaceutical companies are developing slow-release forms of MMF in order to improve its bioavailability and reduce the side effects associated to DMF
[80]. Thus, Alkernes has developed ALK8700 (diroximel fumarate,
12, ), an MMF prodrug with reduced side effects that is now in phase III trials for MS
[97]. Tepilamide fumarate (XP23829,
13, ) is another MMF prodrug, which has been developed by XenoPort
[18]. This compound has shown better solubility and permeability compared to DMF, and also an improved efficacy and reduced gastrointestinal side effects, and it is now in phase II clinical trials for the treatment of plaque psoriasis
[18]. A conjugate of MMF and docosahexaenoic acid (CAT4001) is being developed by Catabasis Pharmaceuticals. In animal models, it has shown promising activities for the treatment of NDDs, such as FRDA and ALS
[18]. With a similar design, V ClinBio developed conjugates built around central glycerol unit and containing two molecules of MMF linked to one of eicosapentenoic acid (VCB101,
14a, ) or docosahexenoic acid (VCB102,
14b, ) for the treatment of MS and psoriasis
[18].
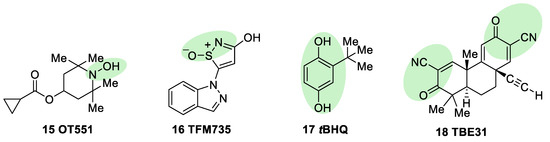
OT551 (
15, ) is an
N,N-disubstituted hydroxylamine developed by Othera Pharmaceuticals
[7][98]. It is a prodrug whose active metabolite TEMPOL (4-hydroxy TEMPO, 4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl) inhibits oxidative stress and disease-associated inflammation by a complex mechanism that, besides its radical scavenging activity, involves a reduced activation of NF-κB
[99] in acute inflammation and activation of the KEAP1–NRF2 pathway, both indirectly and by targeting KEAP1
[100]. It was designed for topical use and protected the retinal pigment epithelium and photoreceptors from oxidative damage and inflammation in preclinical studies. It has also shown efficacy in a phase II clinical trial on age-related macular degeneration
[18].
Figure 7. Chemical structures of OT551, TFM735, tBHQ, and TBE31. Their electrophilic moieties or promoieties are highlighted in green.
During a high-throughput screening study, TFM735 (
16, ) was identified as an activator of NRF2 through a Cys151-dependent mechanism
[101]. This compound is currently in preclinical development by Mochida Pharmaceuticals for the treatment of MS
[18].
tBHQ,
tert-butylhydroquinone (
17, ) is a metabolic precursor of an electrophilic quinone that is able to disrupt the KEAP1/NRF2 complex. It is widely used as a food preservative and due to its antioxidant properties, it has been also employed with neuroprotective purposes. Treatment with
tBHQ reduced oxidative stress, and it also prevented neuronal toxicity and Aβ formation in NT2N cell lines
[102]. Furthermore, it also led to a decrease in secondary injury and an improvement in function recovery after traumatic brain injury
[103].
TBE-31 (
18, ), a fully synthetic acetylenic tricyclic bis(cyanoenone), is one of the most potent known NRF2 inducers
[59]. It has been mainly studied in cancer models, presenting strong antioxidant and anti-inflammatory activities. Furthermore, treatment with TBE-31 improved mitochondrial function and protection against oxidative stress in fibroblasts and cerebellar granule neurons in FRDA models
[104].
 +1 point
+1 point