1000/1000
Hot
Most Recent
 +1 point
+1 point
Molecularly imprinted polymers (MIP) are obtained by initiating the polymerization of functional monomers surrounding the template molecule in the presence of crosslinkers and porogens. Usually the best adsorption performance can be obtained by optimizing the polymerization conditions, but the process is time-consuming and labor-intensive. At the same time, the use of a large number of organic reagents in the process of experimental optimization also limits the development and promotion of molecular imprinting technology. Theoretical calculation based on calculation simulation and intermolecular force is an effective method to solve this problem because it is convenient, versatile, environmentally friendly and low in price. It is not affected by the space environment, and the calculation efficiency is high.
Molecularly imprinted polymers (MIPs) are porous materials with specific recognition capacity towards the template molecule, which are obtained by self-assembly of template molecules and functional monomers in a porogen, and then polymerization is initiated in the presence of a cross-linking agent. The process of preparing MIPs is outlined in Figure 1. When the template molecule interacts with the functional monomer, the imprinting site is memorized through multiple action effects and fixed through the polymerization process. After the template is removed, the adsorption cavity complementary in shape and structure to the template molecule is left in the polymer matrix, which can selectively recognize the target molecule. Molecular imprinting technology originated from antibody immunology, that is, the specific combination of “lock and key” between antibody and antigen [1]. In 1973, Wulff [2] prepared organic MIPs for the first time. Since then, MIPs have attracted widespread attention. At present, MIPs, as a kind of intelligent adsorption material, are widely used in various fields, such as chromatographic separation [3], solid phase extraction [4][5][6], sensors [7][8][9], and biomedicine [10][11]. In the past two decades, great progress in MIPs has been achieved (Figure 2). A variety of novel and interesting imprinted polymers, including supramolecular imprinted polymers [12][13], multitemplate imprinted polymers [14][15], multifunctional monomer imprinted polymers [16][17], dummy template imprinted polymers [18][19], and chiral recognition polymers [20][21], have been developed. In fact, synthesis parameters have been obtained through experimental optimization in most cases. Finding complex and cumbersome conditions is time consuming and laborious. Moreover, numerous organic reagents are used. These factors severely restrict the application and promotion of molecular imprinting technology.
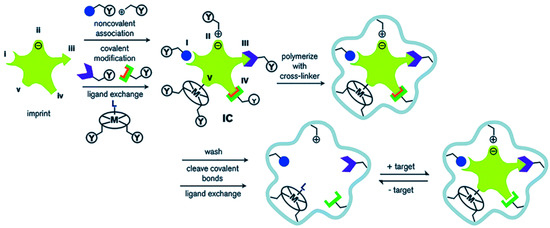
Figure 1. Schematic diagram of the molecular imprinting process: (I) non-covalent, (II) electrostatic/ionic, (III) covalent, (IV) semi-covalent, and (V) coordination to a metal center (Reprinted with permission from [22]. Copyright 2014 Royal Society of Chemistry).
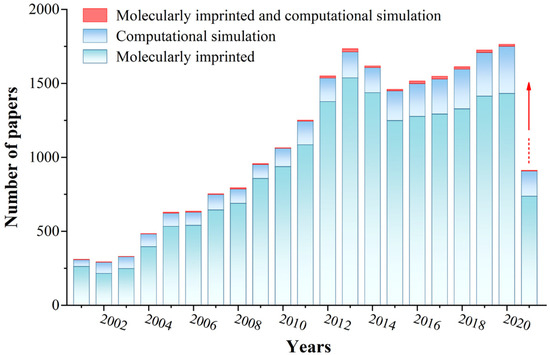
Figure 2. The literature statistics of MIPs and computational simulation. (Database: Scifinder; Search keywords: molecularly imprinted, computational simulation, molecularly imprinted and computational simulation, respectively. Search time: 13 June 2021).
The methods used in the theoretical calculation and simulation of various MIP designs are molecular mechanics (MM), molecular dynamics (MD), and quantum mechanics (QM). The computational cost of MM optimization is considerably lower than that of QM, and thus it is orders of magnitude faster than the latter. However, the accuracy of MM results is limited by simplified calculation models, which allow the reduction in calculation costs. The QM approach can better solve the problem of choosing the appropriate initial direction of interacting molecules because it is more accurate than the other methods. However, the computational complexity of the QM approach exponentially increases as the number of molecules involved in the calculation system increases. The MD method can effectively address this problem. When simulating the dynamic process of the interaction between molecules, changes in the molecule itself are often not considered, thereby making the calculation of the simulation method more efficient. Therefore, the MD method is most widely used when numerous molecules are involved in designing MIPs, such as in optimizing the ratio of template, monomer, and cross-linking agent. The application of MM, MD, and QM methods in MIP simulation is given in Table 1.
Table 1. Theoretical simulation calculation methods for the design of MIPs.
|
Simulation Method |
Template |
Force Field/Method |
Software |
MIPs Design |
|---|---|---|---|---|
|
Molecular mechanics (MM) |
Myoglobin [29] |
OPLS3 |
Prime |
Screening functional monomers |
|
Morphine [30] |
CHARMM and MMFF94 |
Discovery Studio |
Template-monomer ratio |
|
|
AMBER MM |
SPSS Statistics |
Screening functional monomers/template-monomer ratio |
||
|
Norfloxacin [33] |
MMFF94X |
Discovery Studio |
Screening functional monomers/template-monomer ratio |
|
|
Molecular dynamics (MD) |
Curcumin [34], fenthion [35], N-3-oxo-dodecanoyl-L-homoserine lactone [36], methidathion [37], endotoxins [38], phosmet insecticide [39], cocaine [40], methyl parathion [41], aflatoxin B1 [42] |
Tripos |
SYBYL |
Screening functional monomers/template-monomer ratio |
|
Bisphenol A [43], carbamazepine [44], phthalates [45], norfloxacin [46], sulfamethoxazole [47] |
COMPASS |
Materials Studio/accelrys.com |
Screening functional monomers/template-monomer ratio |
|
|
Thiamethoxam [48] |
AMBER |
Gaussian |
Template-monomer ratio and solvent |
|
|
Rhodamine B [49] |
GROMOS |
GROMACS |
Template-monomer ratio and solvent |
|
|
Quantum mechanics (QM) |
Vancomycin [50], primaquine [51], tramadol [52], thiamethoxam [48], clenbuterol [53], sulfadimidine [54], bilobalide [55], chloramphenicol [56], paclitaxel [57], acetamiprid [58], acetazolamide [59], lamotrigine [60], cyanazine [61], 3-methylindole [62], polybrominated diphenyl ethers [63], pirimicarb [64], metoprolol [65], ciprofloxacin or norfloxacin [66] |
DFT |
Gaussian |
Screening functional monomers/template-monomer ratio/solvent |
|
Aspartame [67], pinacolyl methylphosphonate [68], metolachlor deschloro [31], metsulfuron-methyl [32], thiocarbohydrazide [69] |
Semiempirical method |
Spartan/SPSS Statistics |
Screening functional monomers/template-monomer ratio |
|
|
Benzo[a]pyrene [70], tryptophan [71], furosemide [72], buprenorphine [73], hydroxyzine and cetirizine [74], atenolol [75], diazepam [76], metolachlor deschloro [31], metsulfuron-methyl [32], allopurinol [77], methadone [78], clonazepam [79], theophylline [80], ametryn [81], mosapride citrate [82], baicalein [83], |
Ab initio |
HyperChem/Gaussian/AutoDockTools/SPSS Statistics |
Screening functional monomers/template-monomer ratio |
The application of theoretical calculations in designing MIPs is primarily achieved by theoretical simulations and selection of appropriate functional monomers, template molecules, crosslinkers, and their ratios. The binding energy (i.e., electronic interaction energy) between the template molecule and the functional monomer can be simulated and calculated provided that the binding energy between the template molecule and the functional monomer is high, indicating that the corresponding MIPs have excellent selectivity and adsorption performance. In addition, the ratio of the molecular and monomer system is closely related to the imprint factor of MIPs. In general, this ratio is calculated and optimized by performing the computational simulation in a vacuum environment to obtain the Eqaution (1) for the binding energy between the template molecule and the functional monomer.
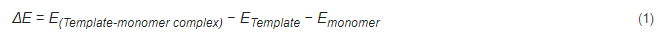
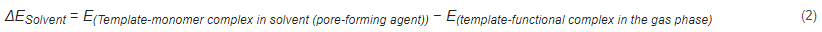
where ΔESolvent is the energy difference between a template molecule and a functional monomer in solution and in a vacuum environment. A weak influence of the solution on noncovalent interactions during molecular fingerprint polymerization results in a small energy difference value, suggesting that the solvent is the best polymerization solvent for obtaining molecular fingerprint polymers [84][85].
Computer molecular modeling technology has been applied to the screening and optimization of molecules in many materials, and it is also a feasible method for preliminary exploration of MIP. Computer simulation reduces the time and reagent-related costs required to obtain the appropriate MIP adsorbent, and significantly reduces the consumption of organic solvents. In addition, it can explain the specific recognition mechanism of imprinted materials at the molecular level. For all the above reasons, the use of computer molecular simulation to design MIP adsorbents in analytical practice not only conforms to the principles of green analytical chemistry, but also explains the nature of MIPs binding to target molecules from the intermolecular forces. The QM method, compared with other methods, can ensure more accurate simulation results in the MIP system dominated by non-bonding interaction, because the smallest structural unit electron was studied and the quantum effect was considered in the method. Therefore, the QM method is also the most widely used in MIP simulation operations. However, in the simulation of macromolecules and polyatomic systems, this method is very time-consuming and even prone to errors. MM and MD are classical mechanics methods. Their smallest structural unit is no longer an electron but an atom. Therefore, the simulation operation complexity of the imprinting system is greatly reduced, and the operation speed is faster. MM method directly utilizes the potential function to study the problem, without considering the kinetic energy and the corresponding structure of the atom. However, the MD method focuses on the movement of atoms in the MIP system and establishes the relationship between temperature and time, which can simulate the imprinting system more realistically, and the simulation results are more representative. In general, the DFT procedure in the QM method was recommended in the MIPs design and mechanism interpretation simulation calculation. However, this also means that the computational complexity of this method increases dramatically for large molecules and systems with a large number of molecules. MD method may be the best solution at this situation, simulated annealing process in particular, which can complete the lowest energy conformation search in a very short time. At present, an increasing number of research have been using multiple calculation methods to achieve complementary advantages when designing and optimizing the experimental parameters of MIPs preparation, so as to ensure more efficient and accurate simulation results. In addition, simulation is also the direction of current efforts. A more realistic simulation environment can make the calculation results accurate and reliable.





