Proteins are fundamental biomolecules of living cells, and their expression levels depend on the balance between the synthesis and degradation. The genetic manipulation of the target protein using CRISPR/Cas9, Cre/loxP, tetracyclin system, and RNA interference, are widely used for the regulation of proteins at the DNA, transcriptional, or mRNA level. Recently, researchers have developed various types of molecular tools for the regulation of protein expression at the post-translational level, which rely on harnessing cellular proteolytic machinery including ubiquitin–proteasome pathway, autophagy-lysosome pathway, and endocytosis. The post-translational control of protein expression using small molecules, antibodies, and light can offer significant advantages regarding speed, tunability, and reversibility. These technologies are expected to be applied to pharmacotherapy and cell therapy, as well as research tools for fundamental biological studies.
1. Introduction
The manipulation of cellular functions is one of the major approaches in biological research and is supported by the control of biomolecules, including DNA, RNA, proteins, peptides, and bioactive small molecules. In particular, researchers often aim to control the activity of specific proteins in mammalian cells, to understand phenomena of interest. The observation of the cellular output and phenotypes in cases of selective enhancement or perturbation of the activity of the target protein enables the deduction of its function. For this purpose, a variety of methods
[1] are now available that allow the manipulation of the cellular abundance, localization, conformational change, enzymatic activity, and post-translational modification of proteins, as well as their interaction with partners. Among them, controlling protein abundance is a robust approach compared with other methods because it directly ensures activity in cells.
Altering the gene or transcript encoding the target protein is broadly used for the regulation of cellular protein abundance. The CRISPR/Cas9 system, which is one of the representative approaches in this setting, uses a Cas9 endonuclease and a single-guide RNA (sgRNA) with a complementary sequence to the target DNA
[2][3][4]. The sgRNA guides the Cas9 to the target site in the genome, for cleavage. The CRISPR/Cas9 system allows for the precise and efficient deletion or insertion of the target DNA. The Cre/loxP system is another classical technique that also controls protein expression at the DNA level
[5][6][7]. Cre recombinase specifically excises a DNA sequence flanked by two loxP sites. The Cre/loxP system allows the excision from the genome of a target DNA flanked by the loxP sites by expressing Cre proteins in a target tissue and at the desired time. Although the techniques of genetic depletion have greatly contributed to the advancement of biological studies, some concerns remain: first, it is not easy to apply them to the regulation of core-essential genes, which are indispensable for the viability of mammalian cells
[8][9]. Second, the living organism might adapt to the circumstances generated by the genetic depletion
[10], which may mislead the interpretation of the phenotypic observations. Rather than controlling the target molecule at the DNA level, alternative approaches rely on the modulation of the gene of interest at the transcriptional level. The tetracycline (tet) system is a well-known tool for the transcriptional control of target protein expression that can be turned on or off using tet or its derivative, doxycycline (Dox)
[11][12][13]. The tet system uses a fusion protein of the tetracycline ligand-binding domain to a sequence-specific transcription activation domain and a construct that contains a target gene under the control of a minimal promoter sequence combined with tet operator sequences. Another example of transcriptional control is the knockdown method based on RNA interference (RNAi), which is a regulatory mechanism present in most eukaryotic cells. RNAi uses small double-stranded RNA (dsRNA) molecules to direct homology-dependent degradation of the target mRNA
[14][15]. Although these techniques have been widely used as gold standards for the modulation of target proteins, these approaches face a fundamental limitation, i.e., waiting for the natural clearance of the target proteins from the cell
[16]. In the past few decades, researchers have developed technologies that regulate protein expression at the post-translational level. Those technologies enable the fast clearance or stabilization of the target protein, because the stability of the target protein is controlled directly. Most technologies used for post-translational control of protein expression take advantage of cellular proteolytic pathways.
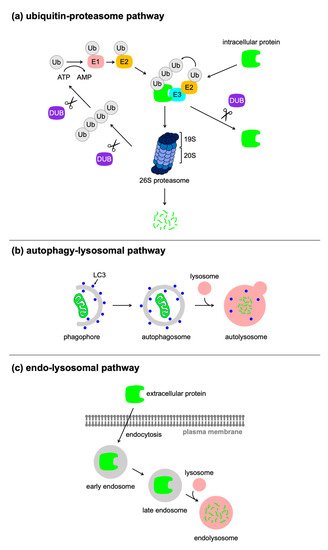
Eukaryotic cells have two major proteolytic pathways: the ubiquitin–proteasome pathway
[17] and the lysosomal pathway
[18]. Many techniques used for the post-translational control of protein expression rely on the ubiquitin–proteasome pathway, which achieves selective proteolysis. Proteolytic substrates are modified with ubiquitin (Ub), a highly conserved 76-amino-acid polypeptide, which is conjugated to an internal lysine of the substrate through an isopeptide bond to the C-terminal glycine of Ub. This reaction is generally called ubiquitination and occurs sequentially through the three steps with activating (E1), conjugating (E2), and ligase (E3) enzymes (
Figure 1a). Ub is initially activated in an ATP-consuming reaction by an E1 enzyme, to which Ub is conjugated by a high-energy thioester bond. Subsequently, the activated Ub is transferred to a cysteine residue of an E2 enzyme, which then catalyzes the transfer of (poly)Ub to the substrate bound to an E3 enzyme. There are two major classes of E3 enzymes, characterized by Really Interesting New Gene (RING) and Homologous to E6-AP Carboxy Terminus (HECT) domains. RING E3 enzymes serve as a hub for the assembly of the E2 enzyme and the substrate, and catalyze the transfer of Ub directly from the E2 enzyme to the substrate. In contrast, HECT E3 enzymes have a distinct function of Ub transfer. The thioester moiety of the complex of activated Ub and the E2 enzyme is reacted with a cysteine residue in the HECT domain of the E3 enzyme, and is finally transferred to the substrate. Seven lysine residues of Ub (K6, K11, K27, K29, K33, K48, and K63) allow the generation of Ub chains via an isopeptide bond between a lysine of one Ub and the C-terminal glycine of another Ub. The ubiquitinated substrate binds to internal Ub receptors in the 19S regulatory complex of the 26S proteasome, which is a multicatalytic protease. The substrate protein bound to the proteasome is unfolded by ATPases and the polyubiquitin chain is removed from the substrate by proteasome-associated deubiquitinating enzymes (DUBs)
[19]. The unfolded protein is translocated into the central proteolytic chamber of 20S proteasome, where it is cleaved into short peptides. Certain types of DUBs are resident in the cytosol and act in the removal of the (poly)ubiquitin from substrates, which stabilizes the protein.
Figure 1. Cellular proteolytic pathways. Eukaryotic cells have several protein degradation pathways: (a) the ubiquitin–proteasome pathway, (b) the autophagy–lysosomal pathway, and (c) the endo-lysosomal pathway.
2. Targeted Protein Degradation Using Bifunctional Molecules or Antibodies
The representative strategies for post-translational control of protein expression are based on the use of bifunctional molecules. Those molecules enable the recruitment of a protein of interest (POI) to the molecular components of the proteolytic pathways, resulting in the selective degradation of POI. Antibodies-based approaches have also been developed. The technologies discussed below are summarized in Table 1.
2.1. PROTAC
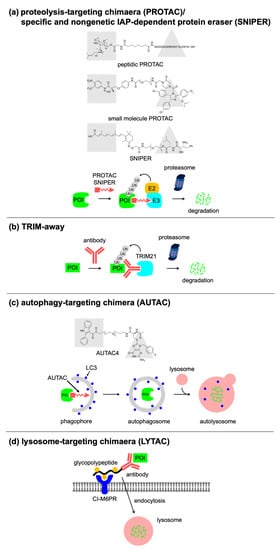
A pioneering work on the targeted protein degradation is the recruitment of the target protein to specific E3 Ub ligases using bifunctional molecules. As a proof-of-concept, the Crews group developed a proteolysis-targeting chimeric molecule (PROTAC) consisting of specific ligands for a protein of interest (POI) and an E3 Ub ligase, with a linker connecting them (
Figure 2a)
[20]. PROTAC binds to both the POI and an E3 Ub ligase, resulting in the ubiquitination, and degradation of the POI. Those authors synthesized a chimeric large molecule called Protac-1 that recruits an E3 Ub ligase complex, Skp1–Cullin-F-box (SCF), to a target protein, methionine aminopeptidase-2 (MetAP-2), resulting in its ubiquitination, and degradation. They further developed an all-small-molecule and cell-permeable PROTAC that consists of a ligand for the non-steroidal androgen receptor and a ligand for the E3 Ub ligase MDM2, connected by a polyethylene glycol (PEG)-based linker (
Figure 2a)
[21]. Itoh et al. developed another chimeric molecule called specific and nongenetic IAP-dependent protein eraser (SNIPER), which recruits inhibitor of the apoptosis protein (IAP) Ub ligases to specifically degrade target proteins
[22].
Figure 2. Targeted protein degradation using molecules or antibodies. Concepts of (a) PROTAC/SNIPER, (b) TRIM-away, (c) AUTAC, and (d) LYTAC.
Such chimeric molecules require significant linker optimization and have a high molecular weight of 900–1100 Da, which limits cellular permeability and solubility. Kihlberg’s group reported solution conformations of a von Hippel–Lindau (VHL) E3 Ub ligase-recruiting PROTAC that has a flexible, PEG-based linker
[23]. This molecule exhibits an elongated conformation in a polar environment while the conformation is folded in an apolar environment. The folded conformation minimizes the size and polarity, allowing it to permeate the cell membrane. Furthermore, a new approach to solve the problem regarding molecular weight of PROTACs has also emerged. Lebraud et al. addressed this by using the bio-orthogonal reaction between two smaller precursors of chimeric molecule, which can occur intracellularly
[24]. They designed a tetrazine-tagged ligand for an E3 Ub ligase that reacts rapidly with a
trans-cyclo-octene–tagged ligand for POI in cells, to form an E3 Ub ligase and recruit PROTAC molecule: termed click-formed proteolysis targeting chimera (CLIPTAC). Recent studies achieved another important improvement of PROTAC using optochemistry technologies. Liu et al. developed a light-inducible switch on PROTACs, termed opto-PROTAC by decorating the ligand moiety for E3 ligase with a photolabile caging group
[25]. This caged compound shows no activity in the dark, whereas restricted degradation can be induced at a specific time and rate after uncaging of photolabile group by light irradiation. Reynders et al. also developed photochemically targeting chimeras (PHOTACs) by introducing azobenzene moieties between ligands for the E3 Ub ligase and the POI
[26]. Light irradiation causes the photoreversible isomerization of the azobenzene moiety and switches the active state to induce protein degradation activity. These technologies are useful for application in clinical settings because the spatiotemporal regulation afforded by them avoids the uncontrolled degradation that causes systemic toxicity.
Table 1. Comparison of the bifunctional molecule-based technologies.
|
Technology
|
Proteolytic Pathway
|
Feature
|
References
|
|
PROTAC/SNIPER
|
Ubiquitin–proteasome pathway
|
Originally developed bifunctional molecules
|
[20][21][22]
|
|
CLIPTAC
|
Ubiquitin–proteasome pathway
|
In-cell generation of bifunctional molecule by the biorthogonal reaction
|
[24]
|
|
opto-PROTAC
|
Ubiquitin–proteasome pathway
|
Light-inducible switch on PROTAC by photolabile caging group
|
[25]
|
|
PHOTAC
|
Ubiquitin–proteasome pathway
|
Light-inducible switch on PROTAC by photo-reversible isomerization of azobenzene moiety
|
[26]
|
|
AbTAC
|
Ubiquitin–proteasome pathway
|
Capable of degrading the cell-surface protein
|
[27]
|
|
Antibody-PROTAC conjugate
|
Ubiquitin–proteasome pathway
|
Cell-type selective targeting
|
[28]
|
|
TRIM away
|
Ubiquitin–proteasome pathway
|
Selective degradation of post-translational modified protein and mutant variants
|
[29][30]
|
|
AUTAC
|
Autophagy-lysosome pathway
|
Capable of targeting cellular organelles
|
[31]
|
|
ATTEC
|
Autophagy-lysosome pathway
|
Molecular glue type degrader for targeting abnormal proteins
|
[32][33]
|
|
LYTAC
|
Endocytosis
|
Capable of targeting extracellular and membrane-bound protein
|
[34][35]
|
2.2. Antibody-Based Approaches
Not only small molecules, but also antibodies are applicable to this type of strategy. Cotton et al. developed antibody-based PROTACs (AbTACs),which facilitates the recruitment of membrane-bound E3 Ub ligases to the targeted cell-surface protein, such as the programmed death-ligand 1 (PD-L1)
[27]. Maneiro et al. proposed a different approach by conjugating the PROTAC molecule with an antibody against the human epidermal receptor 2 (HER2), to selectively target HER2-positive cells
[28]. Once the antibody is recognized by HER2 on the cell-surface, the antibody–PROTAC conjugates are delivered to the cytosol by endosomal internalization. Subsequently, the released PROTAC molecules degrade the cellular target. Another antibody-based approach for targeted protein degradation was reported by Clift et al. They focused on TRIM21, which was originally found to act as an E3 Ub ligase in the immune response to pathogen infection. TRIM21 recognizes antibody-bound pathogens through a high-affinity interaction with the Fc domain of the antibody and tags the antibody-bound pathogens for ubiquitination and degradation. Clift et al. repurposed TRIM21 to establish a method called Trim-Away that targets endogenous proteins for proteasomal degradation using endogenous or exogenous TRIM21 and antibodies that bind to the POI (
Figure 2b)
[29][30]. Notably, this technique enables the selective degradation of post-translationally modified protein and splice or mutant protein variants, by using a specific antibody, while preserving the unmodified/wild-type protein.
2.3. Autophagy/Lysosome-Based Approaches
Recently, several groups reported alternative approaches using proteolytic machinery including the autophagy–lysosome pathway, which is capable of targeting not only proteins, but also organelles. Arimoto and coworkers found that
S-guanylation targets substrates for K63-linked polyubiquitination and selective autophagy
[31]. They developed autophagy-targeting chimeras (AUTACs) consisting of guanine derivatives that mimic the function of endogenous protein
S-guanylation and a specific ligand for a POI (
Figure 2c). AUTAC targets the POI for selective autophagy, and mitochondria-targeted AUTAC allows the removal of dysfunctional fragmented mitochondria via mitophagy. In contrast, Lu and colleagues developed a distinct strategy that uses molecular glues, i.e., autophagosome-tethering compounds (ATTECs)
[32][33]. By performing small-molecule-microarray-based screening, they identified compounds that bind to both the autophagosome-resident LC3 protein and the mutant huntingtin protein (mHTT), but not with the wild-type HTT protein. mHTT causes Huntington’s disease, which is an incurable neurodegenerative disorder. Besides mHTT, ATTEC can target another disease-causing protein with the expanded polyglutamine stretch, including ataxin-3, and reduce its level.
The Bertozzi group developed lysosome-targeting chimeras (LYTACs) that direct extracellular and membrane proteins for degradation via the endolysosomal pathway. They designed and synthesized LYTACs that consist of a small-molecule or an antibody that binds to a POI and a chemically synthesized glycopeptide ligand that binds to the cation-independent mannose-6-phosphate receptor (CI-M6PR), which is a cell-surface lysosome-shuttling receptor (
Figure 2d)
[34]. LYTAC recruits the POI on the cell-surface to CI-M6PR and triggers endocytosis, followed by lysosomal degradation. Although CI-M6PR is ubiquitously expressed in various tissues, there are other classes of lysosome-trafficking receptors that show a tissue-specific expression pattern, inspiring further research to develop a second class of LYTAC technology. The same group focused on the asialoglycoprotein receptor, a liver-specific lysosome-targeting receptor, which induces clathrin-mediated endocytosis
[35]. They conjugated binders of the POI to a triantenerrary
N-acetylgalactosamine (tri-GalNAc) motif that engages the asialoglycoprotein receptor to induce the degradation of the target protein. This GalNAc–LYTAC technology achieves tissue-specific degradation, which could be beneficial for future therapeutic applications.
3. Conditional Control of Protein Stability Using Genetic Manipulation
Another strategy for the post-translational control of protein expression is the genetic conjugation of a degradation tag to the target protein. A degron is a minimal sequence within a protein that can be targeted by a proteolytic machinery
[36]. The degron is transferable; thus, its genetic fusion to other proteins confers instability to the entire fusion protein. The technologies described below are summarized in
Table 2.
3.1. Small-Molecule-Switchable Degrons
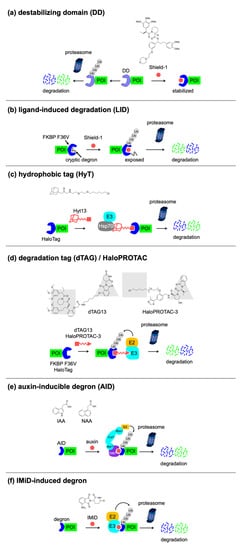
The representative work of small-molecule-based degron technology is the destabilizing domain (DD) developed by the Wandless group. An engineered variant of the human FK506- and rapamycin-binding protein (FKBP12), FKBP12 F36V, possesses a “hole” that allows a “bumped” ligand, Shield-1, to bind more tightly to the F36V mutant compared with the wild-type protein, by almost three orders of magnitude. Using random mutagenesis, they identified mutants of FKBP12 F36V that are unfolded, ubiquitinated, and rapidly and constitutively degraded when expressed in mouse and human-derived cells
[37]. These domains are called destabilizing domains (DDs), and this instability is conferred to other proteins fused to these DDs. The addition of Shield-1, which binds to the FKBP12 DD and stabilizes the DD protein fold, rescues them from degradation, allowing fusion proteins to perform their functions (
Figure 3a). This “drug-on” system is reversible and tunable, as well as useful for the study of constitutively active enzymes. Those authors further created orthogonal DDs using a combination of
Escherichia coli dihydrofolate reductase
[38], the human estrogen receptor ligand-binding domain
[39], or the bilirubin-binding protein (UnaG)
[40], and their ligands. In addition to reversibility and tunability, the drug-dependent stabilization of the target protein is an important feature of the DD technology. It has been shown that the DD approach works in several organisms
[41][42][43][44][45][46]. Although this is useful for the study of constitutively active enzymes, etc., it requires the continuous exposure of the stabilizing ligand to the cells, for protein expression. The authors established a complementary technique with a ligand-induced degradation (LID) domain in which a 19-amino-acid degron is fused to the C terminus of FKBP12 F36V (
Figure 3b)
[47]. In the absence of Shield-1, the degron is bound to FKBP12 F36V and the fusion protein is stable. In turn, the addition of Shield-1, which binds to FKBP12 F36V and displaces the degron, induces the degradation of the fusion protein.
Figure 3. Small-molecule-switchable degrons. Concepts of (a) DD, (b) LID, (c) HyT, (d) dTAG/HaloPROTAC, (e) AID, and (f) IMiD-induced degron.
3.2. Light-Switchable Degrons
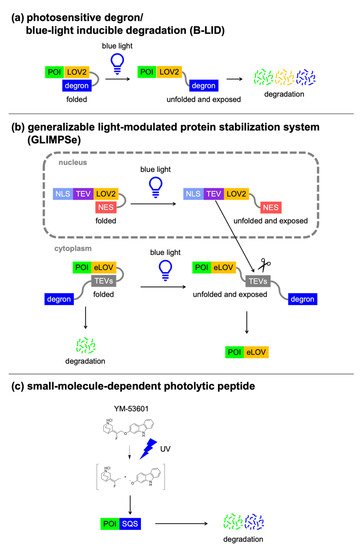
Alternatively, degron activity can be controlled by light irradiation. The light oxygen voltage (LOV2) domain of phototropin consists of a flavin mononucleotide-binding core domain and the C-terminal Jα-helix. Blue light irradiation causes a covalent reaction between a cysteine of the core domain and the flavin mononucleotide (FMN), and induces a conformational change within the core. Then, the Jα-helix is unfolded and dissociated from the LOV2 domain (
Figure 4a). The LOV2 domain technologies are based on the attachment of the degron sequence to the end of the Ja-helix, which can control protein activity by light. Renicke et al. developed a generic photosensitive degron by fusing the
Arabidopsis thaliana LOV2 domain of phototropin 1 to a degron derived from the murine ornithine decarboxylaselike degradation sequence (ODC) (
Figure 4a)
[48], which can be degraded by the 20S proteasome
[49]. When the photosensitive degron is fused to the C terminus of a POI, the Jα-helix interacts with the LOV2 domain in the dark state, whereas blue light irradiation causes the C-terminal Jα-helix to dissociate from the LOV2 domain, thus revealing the ODC degron and inducing the degradation of the fusion protein. The Wandless group also developed a conditional blue light inducible degradation (B-LID) domain by fusing a mutant of the
Avena sativa LOV2 domain to a small peptide degron, RRRGN (
Figure 4a)
[50].
Figure 4. Light-switchable degrons. Concepts of (a) photosensitive degron and B-LID, (b) generalizable light-modulated protein stabilization system (GLIMPSe), and (c) small-molecule-dependent photolytic peptide.
3.3. Nanobody-Based Degrons
Degradation tools that use specific reagents for the detection of the target protein have been emerging. These methods rely on single-domain-binding proteins, such as the heavy-chain antibodies from
Camelidae sp. (called nanobodies)
[51] or the designed ankyrin repeat proteins (DARPins)
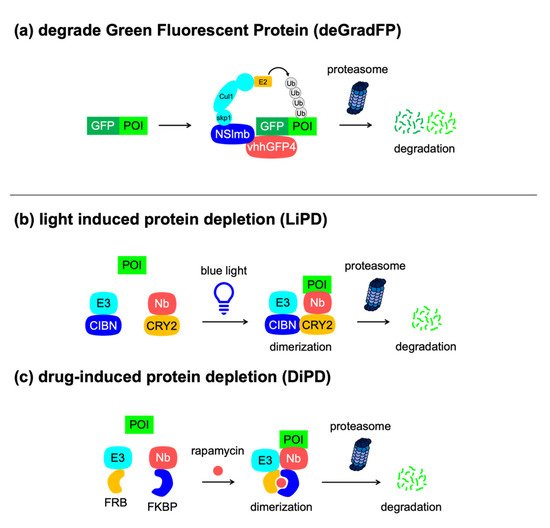
[52]. Unlike antibodies, these single-domain proteins can be expressed by a single vector system, which is useful for biological studies. Caussinus et al. developed a genetically encoded method for the direct and fast depletion of the target green fluorescent protein (GFP), called degrade green fluorescent protein (deGradFP) (
Figure 5a)
[53]. The deGradFP system utilizes a nanobody against GFP, called VhhGFP4, which can be fused with NSlmb, an F-box domain included in the N-terminal part of the F-box protein supernumerary limbs (Slmb) from
D. melanogaster. The engineered NSlmb–VhhGFP protein recruits the SCF complex to the GFP-tagged protein and subsequently ubiquitinates it, thus allowing easy monitoring of the protein removal process. Ludwicki et al. reported a similar approach, named ubiquibody, that uses a fusion protein comprising E3 ligase mimics from a bacterial pathogen and a fibronectin type III monobody that binds to GFP
[54]. This achieves the selective degradation of GFP-tagged proteins in conjunction with hijacking of the mammalian ubiquitin–proteasome pathway. Furthermore, Daniel et al. combined the advantages of the auxin- and nanobody-based degradation technologies to create an AID–VhhGFP fusion for the degradation of GFP-tagged proteins in different cellular structures in a conditional and reversible manner in human cells
[55].
Table 2. Comparison of the technologies used for the conditional control of protein stability using genetic engineering.
|
Degron Technology
|
Tag Size
|
Switch
|
Number of Component(s)
|
Features
|
References
|
|
DD
|
FKBP DD (12 kDa), DHFR DD (18 kDa), etc.
|
Shield-1 (FKBP DD), TMP (DHFR DD), etc.
|
1
|
Drug-induced protein stabilization
|
[37][38][39][40]
|
|
LID
|
FKBP F36V-degron (13 kDa)
|
Shield-1
|
1
|
Drug-induced protein degradation
|
[47]
|
|
HaloPROTAC
|
Halo tag (34 kDa)
|
HaloPROTAC-3
|
1
|
Drug-induced protein degradation
|
[56]
|
|
HyT
|
Halo tag (34 kDa)
|
Hyt13
|
1
|
Drug-induced protein degradation
|
[57]
|
|
dTAG
|
FKBP F36V (12 kDa)
|
dTAG13
|
1
|
Drug-induced protein degradation
|
[58]
|
|
AID
|
AID/IAA (25 kDa), mAID (7 kDa)
|
Auxin (IAA, NAA)
|
2
|
Drug-induced protein degradation
|
[59]
|
|
IMiD-induced degron
|
IKZF3-based degron (6 kDa), SALL4 degron (3 kDa)
|
IMiD (thalidomide, pomalidomide, 5-hydroxythalidomide)
|
1
|
Drug-induced protein degradation
|
[60][61]
|
|
Photosensitive degron/B-LID
|
LOV2-degron (20 kDa)
|
Blue light
|
1
|
Light-induced protein degradation
|
[49][50]
|
|
GLIMPSe
|
eLOV-TEVs-degron (27 kDa)
|
Blue light
|
2
|
Light-induced protein stabilization
|
[62]
|
|
Small-molecule-dependent photolytic peptide
|
SQS C-terminal peptide 371–417 (3 kDa)
|
YM-53601 & UV
|
1
|
Drug- and UV-induced protein degradation, no use of cellular degradation mechanisms
|
[63]
|
|
deGradFP
|
GFP (25 kDa)
|
NA
|
2
|
Protein degradation upon expression of the F-box protein, anti-GFP nanobody, no need to attach tags to POIs
|
[53]
|
|
LiPD
|
NA
|
Blue light
|
2
|
Light-induced protein degradation, no need to attach tags to POIs
|
[64]
|
|
DiPD
|
NA
|
Rapamycin
|
2
|
Drug-induced protein degradation, no need to attach tags to POIs
|
[64]
|
|
SURF
|
3 x FRB degron-Ub-C (58 kDa)
|
Rapamycin
|
2
|
Drug-induced protein stabilization, tag removal via endogenous protease activity
|
[65]
|
|
SMASh
|
SMASh tag (34 kDa)
|
HCV protease inhibitors (ASV, CLV)
|
1
|
Drug-induced protein degradation, tag removal via intramolecular protease activity
|
[66]
|
|
LIBRON
|
FKBP DD-Ub (20 kDa)
DHFR DD-Ub (27 kDa)
|
Shield-1 (FKBP DD-Ub), TMP (DHFR DD-Ub)
|
1
|
Drug-induced protein stabilization, tag removal via endogenous protease activity
|
[67]
|
Figure 5. Nanobody-based degrons. Concepts of (a) deGradFP, (b) LiPD, and (c) DiPD. Nanobody (Nb).
3.4. Excisable Degrons
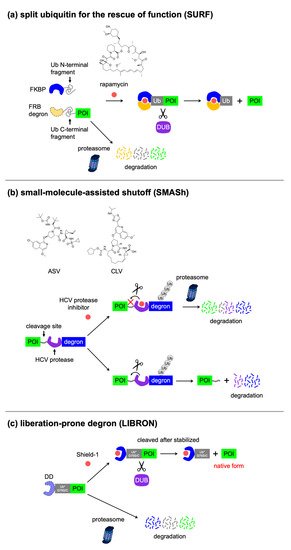
Most of the methods that use the degrons mentioned above, with the exception of LiPD and DiPD, require permanent fusion with degrons at the N or C terminus of the POI. The terminal region of the protein often encodes an important sequence that is indispensable for proper function, such as post-translational modification or interaction with other proteins. This prompted researchers to develop excisable degrons that allow the release of target proteins from degrons. Pratt et al. developed the split Ub for rescue of function (SURF) system
[65], in which cells express a fusion protein of FKBP12 with the Ub N-terminal fragment and a fusion protein of three copies of the FRB mutant W2101F (used as a degron) to the Ub C-terminal fragment, followed by the POI (
Figure 6a). Rapamycin induces the dimerization of FKBP12 and FRB mutant, to allow Ub fragment complementation. The Ub domain is cleaved by cellular DUBs, releasing the POI from the FRB mutant. The SURF system allows the conditional control of protein stability in native from, but requires dimerization of two elements in conjunction with transduction of multiple genes.
Figure 6. Excisable degrons. Concepts of (a) SURF, (b) SMASh, and (c) LIBRON.
 +1 point
+1 point