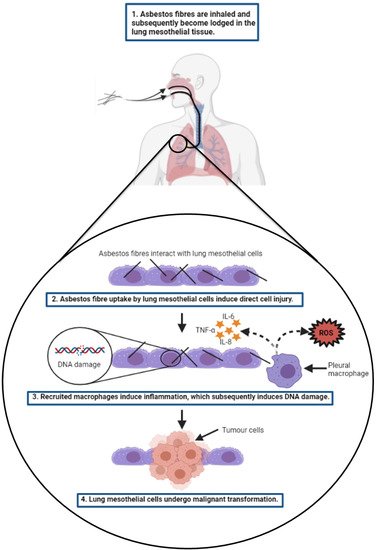
2.3.2. Targeted Therapies
Past research efforts involving a comprehensive genomic profiling of MPM has aided in the detection and characterisation of MPM-specific genetic alterations that represent actionable therapeutic targets for novel MPM treatment strategies. Kinase inhibition, angiogenic inhibition, tumour suppressor genes, and mesothelin are some of the key molecular mechanisms/targets that have been tested for the development of improved MPM treatment strategies.
Receptor tyrosine kinases (RTKs) are a large family of cell cycle regulatory receptors that are commonly over-expressed and activated in MPM
[124]. The activation of these receptors induce biochemical signalling cascades that cause the transduction of aberrant cell growth signalling, which ultimately leads to cancer development and progression. Hence, RTKs represent a potential therapeutic target for MPM and therefore this has attracted research aiming to investigate strategies of RTK inhibition and associated anti-tumour activity. MPM tumours secrete pro-angiogenic factors, such as epidermal growth factor (EGF), platelet-derived growth factor (PDGF), and vascular endothelial growth factor (VEGF), all of which are linked to cancer cell proliferation. High levels of EGF, PDGF and VEGF have been reported in serum and pleural effusions derived from MPM patients, and have been linked to poor prognosis and patient survival
[125][126][127]. The humanised monoclonal antibody against VEGF, bevacizumab, has demonstrated efficacy and been approved for the treatment of several cancers, such as metastatic renal and colorectal cancer, and non-small cell lung cancer. Unfortunately, clinical trials involving MPM patients treated with bevacizumab have not demonstrated a similar efficacy, whereby the addition of bevacizumab to first-line chemotherapy failed to increase the median survival of MPM patients
[128][129][130]; albeit with the exception of a recent phase III trial where bevacizumab-treated patients displayed a modest increase in survival of 2.6 months compared to the standard care treated cohort
[7]. Clinical trials involving the use of the imatinib and dasatinib antibody inhibitors against the PDGF beta-receptor (PDGFRβ), which is known to be over-expressed in mesothelioma cell lines, have similarly yielded disappointing results in regards to patient survival
[131][132][133][134]. Furthermore, inhibitors of EGFR, such as erlotinib and gefitinib, have also failed in clinical trials despite the overexpression of EGFR in MPM specimens
[135][136]. Interestingly, a reduction of angiogenesis and MPM tumour mass and enhanced tumour sensitivity to chemotherapy drug treatment have been demonstrated in the pre-clinical setting, upon the administration of plasminogen activator inhibitor-1 (PAI-1) to mice bearing intrapleural tumours with high VEGF-A expression
[137][138]. Therefore, the use of PAI-1 inhibitors represents a promising therapeutic strategy for MPM that warrants further exploration. Another promising therapeutic strategy is the suppression of MPM tumour growth via the inhibition of fibroblast growth factor receptors (FGFRs). Fibroblast growth factor 2 (FGF2) levels have been shown to correlate to tumour aggressiveness and poor patient survival and its down-regulation has been shown to induce the suppression of mesothelioma cell proliferation without affecting the growth of non-malignant cells
[139]. An impairment of MPM cell proliferation and migration, both in vitro and in vivo, have been demonstrated upon the inhibition of fibroblast growth factor receptor 1 (FGFR1), as well as enhancing tumour response to chemotherapy drug or ionising irradiation
[140]. These studies have therefore demonstrated the potential for the development of novel FGFR-based treatment strategies for MPM and warrant further pre-clinical investigation.
An alternative tyrosine kinase that has been identified as a potential therapeutic target for MPM is the focal adhesion kinase (FAK) protein. FAK, also known as protein tyrosine kinase 2 (PTK2), is located in the cytosol and regulates cellular adhesion, proliferation, migration, and survival; in addition to being critical for cancer stem cell survival and maintenance
[121]. In many cancers, FAK overexpression has been linked to aggressive tumour behaviour and promotes immune evasion, resulting in tumour survival and progression
[141][142][143][144]. Approximately 35–40% of patients with MPM possess mutations in the neurofibromatosis type 2 (
NF2) gene, which encodes the protein Merlin
[145]. Functional merlin normally inhibits FAK activation, however it has been demonstrated that in
NF2-mutated MPM tumour cells, stable overexpression of FAK induces enhanced tumour invasiveness; which decreased significantly upon restoring Merlin expression
[146]. A recognition of the tumour-promoting role of FAK in MPM has prompted the development of FAK small molecule inhibitors and research investigating their potential for use in MPM-based targeted therapies. The small molecule FAK inhibitor, defactinib, yielded promising results in pre-clinical studies in
NF2-deficient MPM in vitro and in vivo
[147], however, it failed to display any enhanced therapeutic benefit in a subsequent phase II clinical trial in MPM patients and consequently the trial was suspended in late 2015
[121]. An alternative FAK inhibitor, PND-1186 (otherwise known as VS-4718), has demonstrated promising efficacy as a potential treatment option for MPM after it was found that it inhibited proliferation and induced apoptosis in MPM cells lacking Merlin expression
[148]. The preferential anti-tumour effect of PND-1186 in Merlin-deficient MPM cells suggests that Merlin is a potential predictive biomarker for an enhanced MPM tumour sensitivity to PND-1186. This postulation is also evidenced by an earlier Phase I clinical trial whereby Merlin-deficient MPM patients treated with the FAK inhibitor, GSK2256098, exhibited an increased progression-free survival (PFS) and disease stabilization
[149]. Additionally, the expression of the tumour-suppressor protein, E-cadherin (CDH1), has been identified as another predictive marker of MPM tumour response to FAK inhibition after a study by Kato et al. revealed that a subset of Merlin-deficient MPM cells with E-cadherin-positive expression exhibited resistance to PND-1186 treatment
[150]. To address the issue of MPM resistance to FAK inhibitor treatment, prospective pre-clinical studies exploring the efficacy of FAK inhibitor treatments in combination with other functional biomolecules, such as tumour suppressor miRNA mimics, are greatly needed. For instance, a previous bioinformatics study of miRNA expression in MPM has indicated that a number of down-regulated tumour-suppressor miRNAs have a strong link to FAK involvement
[151]. The replacement of these down-regulated miRNAs with functional miRNA mimics may potentially be the key to sensitizing E-cadherin-positive MPM to FAK inhibitor treatment.
Tumour suppressor inactivation (i.e., loss-of-function) constitutes one of the most typical mutational events in MPM tumours.
BAP1 and
CDKN2A are tumour suppressor genes that are frequently inactivated in MPM, and have therefore attracted widespread research interest focused on exploiting these altered genes as potential candidates for the development of targeted therapies
[152][153][154][155].
BAP1-mutant MPM cell lines have been shown to be significantly less sensitive than the wild-type counterpart when exposed to the chemotherapy drug, gemcitabine, with significantly less DNA damage being detected in the
BAP1-mutant cells
[156]. Hence, research efforts have focused on exploring treatment options that can potentially sensitise
BAP1-deficient MPM to chemotherapy drug treatment. One particular option that has been investigated is the induction of synthetic lethality of alternate DNA repair pathways, such as through the administration of poly-ADP ribose polymerase (PARP) inhibitors, which have been proven to induce cell death in
BAP1-deficient MPM cell lines
[61][157]. Promisingly, it has recently been established that the
BAP1 mutation status alone does not confer MPM cell sensitivity to PARP inhibition. Rather a combination of both PARP inhibitor and the chemotherapy drug, temozolomide, was found to exhibit efficacy in
BAP1-deficient MPM cell lines with an associated high and low expression of Schlafen 11 and O-6-Methylguanine-DNA Methyltransferase (MGMT), respectively
[158]. Hence, it has been proposed that such a treatment strategy would be ideal for MPM patients whose tumours possess these combined genetic traits. Furthermore, BAP1 inactivation has been considered to play a key role as an epigenetic regulator associated with the polycomb repressive complex 2 (PRC2) and an enhancer of the zeste homolog 2 (EZH2) pathway. As such, MPM with BAP1 loss have been shown to be sensitised to EZH2 inhibition, both in vitro and in vivo
[159]. A phase II trial involving treatment of mesothelioma patients with the EZH2 inhibitor, tazemetostat, demonstrated a promising disease control rate of 51% at 12 weeks
[160].
In addition to
BAP1-targeted strategies, researchers have investigated therapeutic strategies that are specific to the
CDKN2A mutation of MPM tumours.
CDKN2A encodes the ADP-ribosylation factor (ARF, commonly known as p14) and INK4A (commonly known as p16), and function by regulating the expression of the p53, and pRB tumour suppressors; ultimately leading to G1 arrest and G0 arrest/apoptosis, respectively
[120]. The whole
INK4a/ARF locus is typically deleted in more than 70% of human mesothelioma cell lines, which is associated with a loss of function of p53 and pRb, and a consequent failure of cell cycle arrest. It has been shown however, that the restoration of
p14ARF via adenoviral
p14ARF transfection in human mesothelioma cell lines, induces cell cycle arrest, growth inhibition and apoptosis; which was associated with increased p53 levels and dephosphorylation of pRB
[161]. Alternatively, researchers have investigated therapeutic strategies that exploit INK4A and its downstream targets, such as cyclin-dependent kinase 4 (CDK4) and CDK6. The presence of INK4A is associated with an inhibition of CDK4 and CDK6 activity, which leads to cell cycle arrest. In INK4A-deficient MPM cells however, CDK4 and CDK6 remain active and promote cell cycle progression. Therefore researchers have employed small molecule inhibitors of CDK4 and CDK6, such as abemaciclib, to induce apoptosis in MPM tumours and a suppression of tumour growth when used in conjunction with radiation in MPM mouse models
[162]. These promising results have led to a current clinical trial (NCT03654833 (MiST)) investigating the efficacy of abemaciclib in
p16INK4A-deficient MPM patients. The direct restoration of INK4A via gene therapy has also been investigated in pre-clinical models of mesothelioma and has demonstrated promising anti-tumour activity. In particular, the transduction of MPM cells with the INK4A-expressing adenovirus, Adp16, has been shown to effectively induce cell cycle arrest, reduced cell growth, and eventual cell death
[163]. Furthermore, the restoration of INK4A in mesothelioma xenografts was shown to be associated with an inhibition of tumour growth and a reduction in tumour size and spread
[163]. Despite these promising results, the re-expression of INK4A via gene therapy has not yet been tested in clinical trials.
Tumour suppressor miRNA have been investigated for their potential role as an MPM targeted therapy option. As discussed previously, a downregulation of miR-16 in MPM cells and tumours is associated with MPM cell/tumour growth. To address this, a restoration of miR-16-5p was investigated both in vitro and in vivo upon reverse transfecting MPM cells with miR-16-5p mimics and administering miR-16-5p-loaded minicells to MPM xenografted tumours, respectively
[66]. This study demonstrated that the synthetic restoration of miR-16 in the MPM cells inhibited their growth and sensitised them to chemotherapy drug treatment with pemetrexed and gemcitabine. Furthermore, the intravenous administration of the miR-16-5p-loaded minicells to the tumour-bearing mice resulted in a consistent and dose-dependent inhibition of MPM tumour growth. Additionally, the in vitro and in vivo restoration of the tumour suppressor, miR-193a-3p, displayed similar promising results; inducing an inhibition of MPM cell growth and induction of apoptosis and necrosis in vitro, and inhibition of xenograft tumour growth in vivo
[67]. The results from these studies ultimately provided the supportive basis for a phase I clinical trial, MesomiR-1; the first and only in-man miRNA study to date, which investigated the safety and optimal dose of a miR-16-based mimic delivered via anti-EGFR antibody-targeted bacterial minicells, dubbed TargomiR’s. Results generated from this trial validated the safety of the treatment in all 27 patients, with one patient exhibiting an objective response
[164] and stable disease in a further 15 patients
[165]. Given that only one objective response was attained out of the 27 patients, it has been proposed that a combination treatment approach involving miRNAs combined with other treatment modalities (i.e., chemotherapy) may be necessary in order to elicit an enhanced anti-tumour response
[165]. Additionally, the use of alternative miRNA mimics consisting of both two active 5p and 3p arms have been proposed, as well as alternative delivery systems to more accurately deliver the miRNA mimics to MPM tumour cells
[166].
Other targeted therapy studies have investigated tumour-specific antigens as potential therapeutic targets for MPM, such as mesothelin. Mesothelin is typically expressed at low levels in most normal tissue types, however, it is overexpressed in a variety of solid tumour types, including MPM
[41]. The exact biological function of mesothelin overexpression in MPM is not known, however, pre-clinical evidence suggests that mesothelin plays a role in cell adhesion after it was identified as being the receptor for cancer antigen-125 (CA-125); an interaction which causes heterotypic adhesion and facilitates tumour metastasis
[167][168]. For these reasons, mesothelin has been deemed a potential therapeutic target for novel MPM therapies and has attracted considerable research interest. Three different types of mesothelin-targeting agents have been explored, which include anti-tumour antibodies/ antibodies-drug conjugates, mesothelin-targeting vaccines, and mesothelin-targeting recombinant T cells
[41]. In particular, the humanised antibody, amatuximab, has shown encouraging anti-tumour activity in combination with chemotherapy drugs against mesothelin-expressing tumours, via inhibition of the interaction between mesothelin and cancer antigen 125 (CA- 125)
[169][170]. The immunotoxin, SS1P, consisting of the variable fragment of amatuximab linked to the cytotoxic bacterial toxin
Pseudomonas exotoxin A has also exhibited enhanced anti-tumour activity against mesothelin-expressing cancers, including tumour cell lines derived from ascites of patients with peritoneal mesotheliomas
[171][172]. Despite this, SS1P has shown only modest activity and responses in treated patients of phase I clinical trials, which has been attributed to the immune-induced formation of neutralizing antibodies acting on SS1P after the first cycle of treatment. To overcome this associated immunogenicity limitation of SS1P, a less immunogenic version (RG7787) has been developed and tested pre-clinically in mesothelin-expressing cancers
[173][174]. Alternative mesothelin-specific antibody-drug conjugates that have been investigated in pre-clinical models include αMSLN-MMAE and BAY 94-9343; both of which have exhibited enhanced anti-tumour activity in pre-clinical models compared to standard chemotherapy drug treatment
[175][176]. In addition to mesothelin-specific antibodies, genetically modified mesothelin-targeting vaccines have also been investigated for therapeutic potential. In addition to mesothelin-specific antibodies, recent research has focused on vaccine-based targeting of mesothelin-expressing tumour cells. The mesothelin-expressing CRS-207 is a genetically modified
Lysteria monocytogenes attenuated vaccine, which has demonstrated therapeutic efficacy in pancreatic tumour-bearing pre-clinical murine models
[177]. The in vitro and in vivo pre-clinical testing of CRS-207 in MPM is scarcely reported in the literature, however, a recent clinical-based study has revealed the vaccine has promising therapeutic potential in treated MPM patients; resulting in disease control for 89% of patients and 31% showing a reduction in tumour size
[178].
Alternative pre-clinical studies have highlighted argininosuccinate synthetase 1 (ASS1) as a promising targeted therapy candidate for MPM. ASS1 is a rate-limiting enzyme that controls the production of arginine; a known precursor to biomolecules (e.g., nitric oxides and polyamines) that play a role in tumourigenesis
[179][180][181]. A study by Szlosarek et al. established that 63% of MPM patient-derived tissue samples exhibited either reduced or absent ASS1 levels. Furthermore, this study demonstrated that ASS1-negative MPM cell lines showed a marked decline in cell viability upon withdrawal of arginine from the culture medium, thus highlighting the potential role of arginine depletion therapy for ASS1-negative MPM patients
[182]. This led to a phase II multicentre study which assessed the efficacy of a pegylated arginine deiminase (Adi-PEG 20), an arginine-depleting enzyme, in patients with ASS1-negative MPM. The Adi-PEG 20-treated group exhibited a modest increase in median PFS (3.2 months) in comparison to the group without Adi-PEG 20 treatment (2 months)
[183]. Additionally, a phase I dose escalation study demonstrated that the combination of Adi-PEG 20 with cisplatin and pemetrexed was associated with a 78% response rate in a small cohort of ASS1-deficient thoracic cancer patients (including MPM)
[184]. A phase 2/3 clinical trial (NCT02709512) is further investigating the efficacy of Adi-PEG 20 treatment in combination with standard cisplatin-pemetrexed therapy in ASS1-negative MPM patients.
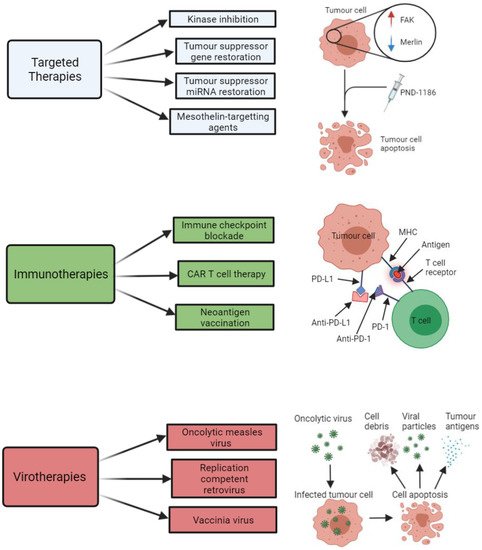
The main types of targeted therapies that have been investigated with respect to treatment development for MPM are summarised below in Figure 2. A summary of some of the key clinical trials that have arisen from pre-clinical targeted therapy-based MPM studies are shown in Table 3.
Figure 2. Summary of the key therapeutic strategies that have been explored by researchers investigating potential new treatment options for malignant pleural mesothelioma (MPM). The three key therapeutic strategies include targeted therapies (e.g., kinase inhibitors, such as PND-1186), immunotherapies (e.g., immune checkpoint inhibitors of PD-L1 and PD-1), and virotherapies (e.g., oncolytic measles virus-mediated tumour cell death). All images were created with
BioRender.com (accessed on 14 September 2021). Abbreviations: FAK, focal adhesion kinase; miRNA, microRNA; CAR T, chimeric antigen receptor T cell; MHC, major histocompatibility complex; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1.
Table 3. Key clinical trials that have arisen from pre-clinical MPM studies.
| Clinical Trial Code |
Phase |
Title |
Treatment |
Target |
Completed (C) or In Progress (IP) |
Outcome |
Reference |
| Targeted Therapies |
| NCT02860286 |
II |
Study of the EZH2 Inhibitor Tazemetostat in Malignant Mesothelioma |
Tazemetostat |
BAP1-deficient MPM |
C |
Disease control was achieved in 51% of patients at 12 weeks and 25% of patients at 24 weeks |
[160] |
| NCT00651456 |
III |
Mesothelioma Avastin Plus Pemetrexed-cisplatin Study (MAPS) |
CT + bevacizumab |
VEGF |
C |
2.6 month increase in patient survival compared to standard CT |
[7] |
| NCT03762018 |
III |
BEAT-meso: Bevacizumab and Atezolizumab in Malignant Pleural Mesothelioma (BEAT-meso) |
CT + bevacizumab + atezolizumab |
VEGF |
IP |
- |
- |
| NCT03654833 |
II |
Mesothelioma Stratified Therapy (MiST): A Multi-drug Phase II Trial in Malignant Mesothelioma (MiST) |
Abemaciclib |
p16INK4A-deficient MPM |
IP |
- |
- |
| NCT02369198 |
I |
MesomiR 1: A Phase I Study of TargomiRs as 2nd or 3rd Line Treatment for Patients with Recurrent MPM and NSCLC |
TargomiRs |
EGFR |
C |
Objective response achieved for 1 patient and stable disease achieved for 15 patients in a cohort of 27 patients |
[164][165] |
| NCT01675765 |
I |
Safety and Efficacy of Listeria in Combination with Chemotherapy as Front-line Treatment for Malignant Pleural Mesothelioma |
CT + CRS-207 vaccine |
Mesothelin |
C |
Disease control achieved in 89% of patients, with 31% showing a reduction in tumour size |
[178] |
| NCT01279967 |
II |
A Clinical Trial of ADI-PEG 20TM in Patients with Malignant Pleural Mesothelioma (ADAM) |
Adi-PEG 20 |
ASS1-deficient MPM |
C |
3.2 months PFS achieved for Adi-PEG 20-treated patients compared to 2 months for patients without Adi-PEG 20 treatment |
[183] |
| NCT02709512 |
II/III |
Ph 2/3 Study in Subjects with MPM to Assess ADI-PEG 20 with Pemetrexed and Cisplatin (ATOMIC) |
CT + Adi-PEG 20 |
ASS1-deficient MPM |
IP |
- |
- |
| Immunotherapies |
| NCT02054806 |
I |
Study of Pembrolizumab (MK-3475) in Participants with Advanced Solid Tumors (MK-3475-028/KEYNOTE-28) |
Pembrolizumab |
PD-1 |
C |
Objective response was achieved in 28% of patients and stable disease achieved in 48% |
[185] |
| NCT02991482 |
III |
Pembrolizumab Immunotherapy Versus Standard Chemotherapy for Advanced Pre-treated Malignant Pleural Mesothelioma (PROMISE-meso) |
CT + pembrolizumab |
PD-1 |
IP |
- |
- |
| NCT02899299 |
III |
Study of Nivolumab Combined with Ipilimumab Versus Pemetrexed and Cisplatin or Carboplatin as First Line Therapy in Unresectable Pleural Mesothelioma Patients (CheckMate743) |
CT + nivolumab + ipilimumab |
PD-1 and CTLA-4 |
IP |
A median overall patient survival of 18 months was achieved for patients treated with CT in combination with nivolumab and ipilimumab, compared to 14 months for patients receiving CT alone |
[123] |
| NCT01722149 |
I |
Re-directed T Cells for the Treatment (FAP)-Positive Malignant Pleural Mesothelioma |
FAP-specific CAR T cells |
FAP |
C |
- |
- |
| Virotherapies |
| NCT01721018 |
I/IIa |
Intrapleural Administration of HSV1716 to Treat Patients with Malignant Pleural Mesothelioma. (1716-12) |
HSV-1716 |
MPM tumour cells |
C |
A median survival of 15 months and 18 months was achieved in HSV-1716-treated patients and HSV-1716-treated patients that exhibited evidence of anti-tumour immunogenicity. |
[186] |
2.3.3. Immunotherapies
There has been growing research interest in immunotherapy as a potential treatment option for MPM after several reported cases of spontaneous regression of MPM identified an associated lymphocyte infiltration in the tumour, which correlated to improved patient survival
[187][188][189]. Hence, recent research has focused on strategies to modulate the immune system in order to enhance or facilitate the efficiency of the immune system to elicit an anti-tumour response. Promising types of emerging immunotherapy strategies for MPM include immune checkpoint blockade (ICPB), chimeric antigen receptor (CAR) T cell therapy, T regulatory cell modulation, and neoantigen vaccination.
It has been established that MPM tumours are capable of evading elimination by the immune system; mediated by the involvement of T-cell inhibitory molecules, such as the cytotoxic T-lymphocyte-associated protein 4 (CLTA-4), programmed cell death protein 1 (PD-1), and PD-L1. PD-L1 is known to be expressed in many cancer cells, including MPM, and is associated with a poorer MPM patient median survival of 5 months as opposed to 14.5 months for MPM patients with tumours lacking PD-L1
[79][80]. The binding of PD-L1 to PD-1 on T-cells impedes immune responses against the tumour by inhibiting T-cell proliferation and activation. Thus, tumours that express PD-L1 are capable of evading cytotoxic T-cell activity. Researchers have therefore focused on methods that aim to intervene and disable the interaction between PD-L1 and PD-1, such as through the use of immune checkpoint inhibitors that are able to block this interaction and enable T- and B-cell re-activation
[190]. Typical immune checkpoint inhibitors include the monoclonal antibodies tremelimumab, pembrolizumab, and avelumab, which act against CTLA-4, PD-1, and PD-L1, respectively. These antibodies have yielded promising clinical results in melanoma and other cancer types
[41][191]. Furthermore, the blockade of the PD-1/PD-L1 interaction and an associated inhibition of tumour growth has been demonstrated in animal models for non-MPM-based studies
[192][193]. Limited pre-clinical data has been published for ICPB in MPM however, although a number of ICPB-based clinical studies for MPM have been conducted. The KEYNOTE-28 trial in particular, whereby MPM patients were treated with pembrolizumab, demonstrated some promising results; with up to 28% of patients exhibiting a beneficial response to the drug and another 48% achieving a stable disease response
[185]. In contrast, one of the most extensive trials for ICPB in MPM, whereby relapsed MPM patients were treated with tremelimumab, reported that 81% of MPM patients died without a significant difference in overall survival between the drug-treated and placebo-treated cohort
[194]. In an effort to improve MPM patient response to ICPB therapy, researchers have resorted to testing the combination of ICPB with other immune-modulatory molecules, targeted therapies, anti-angiogenic agents, chemotherapy drugs, or radiotherapy
[195]. The use of immune checkpoint inhibitors in combination with chemotherapy agents has particularly been shown to have enhanced therapeutic benefit in MPM. This was effectively demonstrated in both pre-clinical models and human subjects upon combining the immune checkpoint inhibitor, anti-PD-1, with the chemotherapy drug, gemcitabine, which resulted in an enhanced tumour control and survival outcome when compared to treatment with either agent alone
[196]. The combination of anti-PD-1 ICPB with other types of chemotherapy agents, such as cisplatin and paclitaxel, have been shown to exhibit enhanced anti-tumour immunity in other thoracic cancer types, commonly inducing a 50% tumour regression, however, their efficacy is yet to be established for MPM
[197]. The use of radiotherapy in combination with ICPB is a promising alternative however, having been investigated in MPM in a recent in vivo study by Wu et al. This study effectively demonstrated a significant inhibition of tumour growth in murine MPM models, which was associated with an increased T cell infiltration into the tumour, upon subjecting them to local radiotherapy in combination with the antibody-mediated blockade of the immune-suppressive cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)
[198]. The use of tumour suppressor miRNA mimics in combination with ICPB also constitutes a potential therapeutic strategy for MPM that warrants investigation. The rationale for this is based on a previous study by Kao et al., which demonstrated that PD-L1 overexpression in a cohort of 72 patients was associated with a downregulation of tumour suppressor miRNAs miR-15b, miR-16, miR-193a-3p, miR-195, and miR-200c
[199]. This same study demonstrated that the treatment of MPM cells with miR-15a and miR-16 mimics led to a downregulation in PD-L1 mRNA and protein levels.
An alternative to the ICPB immunotherapy-based approach that has been explored for MPM is CAR T cell therapy. This form of immunotherapy, based on adoptive cell transfer, involves the generation of T cells that are engineered to recognise specific antigen receptors on the tumour cells. For instance, a study carried out by Klampatsa et al. demonstrated increased cytotoxicity and cytokine release in a panel of four MPM cell lines after patient T-cells were engineered via retroviral transduction to express a panErbB-targeted CAR, co-expressed with a chimeric cytokine receptor in order to promote interleukin-4-mediated CAR T cell proliferation
[200]. Alternative studies have employed mesothelin-targeting recombinant T cells to explore their associated therapeutic potential in MPM in both pre-clinical in vitro and in vivo models, having exhibited robust anti-tumour activity in MPM
[201][202]. An in vivo study effectively demonstrated the potential efficacy of CAR T cell therapy for MPM, whereby an intrapleural mesothelioma mouse model treated with mesothelin-specific CAR T cells injected into the peritoneum resulted in potent and prolonged anti-tumour immunity
[203]. Other targets that have been explored for CAR T cells include components of the stroma of tumours, such as the fibroblast-activating protein (FAP). FAP-specific CAR T cells have exhibited efficacious results in murine subcutaneous MPM models with minimal associated toxicity, which resulted in a phase I clinical trial of human FAP-specific CAR T cells via intrapleural administration in MPM patients
[203].
Other researchers are investigating the potential utility of neoantigen vaccination as a means to stimulate the immune system to initiate an immune-mediated anti-tumour response against MPM. The aim of neoantigen vaccination is to prime the host immune system to recognise and target the foreign MPM tumour cells. Neoantigens arise as a result of mutations to oncogenes and tumour suppressor genes, oncogenic viruses, oncofoetal proteins, or overexpression of proteins
[120]. They constitute attractive candidates for the development of immune-mediated anti-tumour vaccines given that they; (i) exhibit high binding affinities for T cell and human leukocyte antigen (HLA) receptors, (ii) their expression is restricted only to tumour cells, and (iii) their collective binding affinity and specificity effects enable them to bypass central tolerance and issues associated with autoimmunity
[204]. In order to elicit an immune-mediated anti-tumour response, the neoantigens must be presented to T cells upon binding to MHC molecules. Neoantigen vaccination represents a potential form of personalised therapy that is uniquely tailored to individual MPM patients on the basis that their tumour sample can be sequenced using next-generation sequencing (NGS) technology to detect aberrant mutation expression of the neoantigens via RNAseq, and MHC binding potential determined in silico
[120][205]. The capacity to identify MPM tumour-specific neoantigens using NGS platforms and capability of vaccinating patients with their own tumour-specific neoantigens has stimulated considerable research interest in anti-cancer vaccination strategies for MPM. One recent study conducted by Sneddon et al. utilised pleural effusion samples collected from 27 mesothelioma patients to screen and identify potential tumour cell-derived neoantigens. A median value of up to 68 different neoantigens was found to be responsive to CD8+ T cells, with a particularly strong CD8+ T cell response being detected for a predicted neoantigen produced by a spontaneous mutation in the
ROBO3 gene
[206]. This is just one example of a pre-clinical study that provides justified evidence that neoantigens represent promising actionable targets for the development of personalised MPM anti-tumour vaccines. Neoantigen vaccination development for MPM is still in its infancy however, and further pre-clinical research is required to completely elucidate its efficacy and feasibility as a potential new treatment option for MPM patients.
The most common types of immunotherapies that have been investigated in relation to MPM are summarised in Figure 2. A summary of some of the key clinical trials that have arisen from pre-clinical immunotherapy-based MPM studies is shown in Table 3.
 +1 point
+1 point