1000/1000
Hot
Most Recent
 +1 point
+1 point
Guanyl radical or neutral guanine radical G(-H)• results from the loss of a H-atom or an electron/proton (e–/H+) couple from the guanine structures (G). Guanyl radical G(-H)• exists in two tautomeric forms and its role in single- and double-stranded oligonucleotides, in DNA and G-quadruplex, attracted considerable attention since directly connected to the damage of genetic material and its biological consequences. The emerging picture is still incomplete and extrapolation of its chemistry from nucleosides to more complex environment like DNA can be misleading.
The two tautomers of guanyl radical G(-H)• in nucleosides (like guanosine or 2′-deoxyguanosine) are G(N1-H)• and G(N2-H)• after loosing H from N1 and N2 positions, respectively (Figure 1) [1]. The unpaired electron is highly delocalized in both tautomers. The insert of Figure 1 shows that in G(N1-H)• the unpaired electron is mainly localized at the N3, O6, C5, and C8 atoms (spin densities of 0.30, 0.34, 0.21, and 0.27, respectively), whereas in G(N2-H)• the unpaired electron is mainly localized at the N3, C5, C8, and N2 atoms (spin densities of 0.34, 0.29, 0.22, and 0.39, respectively). G(N2-H)• can exist as two conformers in which the remaining N2–H is either syn or anti with respect to the guanine N3 atom (syn-conformer is lower in energy than the anti-conformer). As each tautomer has various resonance forms (discussed in some details in reference [2]), the drawings of G(N1-H)• and G(N2-H)• in Figure 1, where the unpaired electron is placed in the C5 position, are chosen for a facile distinction of the two tautomers. The insert of Figure 1 shows the optical absorption spectra of the two tautomers, the G(N2-H)• being characterized by the absorption band around 620 nm.
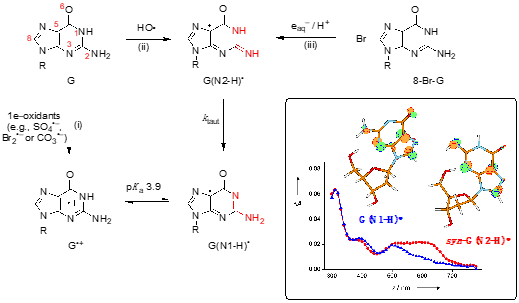
Figure 1. Structure G represents the guanine moiety in guanosine or 2′-deoxyguanosine (the atom numbering of the guanine moiety is given in red). Three pathways are shown for the formation of guanyl radicals G(N1-H)• or G(N2-H)• (the part of the molecule involved in tautomerization is highlighted in red) [1]. Insert: SOMO of G(N1-H)• and syn-G(N2-H)• computed at the B3LYP/6-311G** level and their absorption spectra obtained by pulse radiolysis (taken from ref [2]).
The chemical pathways for the formation of G(N1-H)• or G(N2-H)• are reported in Figure 1: (i) one-electron oxidation of G to give G•+, associated with a pKa value of 3.9, and the deprotonation giving the most stable tautomer G(N1-H)• [3], (ii) the reaction of HO• radical with G proceeding mainly by H-atom abstraction from the exocyclic NH2 group (~65%) to give G(N2-H)• [4][5], and (iii) the one-electron reduction of 8-bromo-guanine derivatives coupled with protonation, giving exclusively G(N2-H)• [2,6]. In the last two pathways tautomerization is observed with a rate constant ktaut = 5 x 104 s–1 at room temperature; the Arrhenius parameters, log(A/s–1) = 8.7 ± 0.4 and Ea = 23.0 ± 2.5 kJ mol–1, the solvent kinetic isotope effect (k(H2O)/k(D2O) = 8.0, and the calculations at the PCM/B1B95/6-31+G**//B1B95/6-31+G** level suggested a complex transition state for the water-assisted tautomerization [1][2][6].
Figure 2 shows the overall mechanism of oxidation of 2′-deoxyguanosine (G) by HO• and CO3•–and their main products (8-oxo-G, Fapy-G, 2Ih, Iz and Z) [7][8]. The two oxidants have some common intermediates generated by different routes, e.g., (N1-H)• and 8-HO-G• radicals. 8-HO-G• is the common precursor of the final products 8-oxo-G and Fapy-G. The Fapy-G formation requires ring-opening followed by 1e– reduction or vice versa (i.e., 1e– reduction followed by ring-opening). The formation of Fapy-G depends on the oxygen concentration and the redox environment. 8-oxo-G, having its reduction potential 0.55 V lower than that of G, undergoes further oxidation in the presence of 1e– oxidants. From oxidation of G under aerobic conditions or in the presence of CO3•–, the products 2Ih, Iz and Z are formed via C5 paths
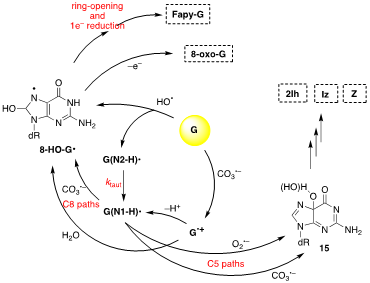
Figure 2. The mechanism of 2′-deoxyguanosine (G) oxidation by hydroxyl radical (HO•) or carbonate radical anion (CO3•–); the final stable products are 8-oxo-G, Fapy-G, 2Ih, Iz and Z.
The mode of formation and the kinetics of decay of G•+ depend strongly on the secondary structures of genetic materials. Single- and double-stranded oligonucleotides (ss-ODNs and ds-ODNs) have been used as model for understanding the behavior of 1e– oxidation of DNA. The 1e– oxidation of ss-ODN like TCGCT behaves similar to 2′-deoxyguanosine [10]. The behavior changes dramatically going to double-stranded substrates.[1]
A large body of experimental data has accumulated during the last 30-years showing that 1e– oxidation of DNA produces a hole that can migrate through the double helix with final destination the G sites[11][12][13]. Among the four common DNA bases (A, G, T and C), G is the most readily oxidized to G radical cation (G•+), which is also the putative initial intermediate in the oxidative DNA damage. Thus, there have been numerous studies of the formation and behavior of G•+ as ds-ODNs using spectroscopic and product studies. Upon formation of G•+ a fast deprotonation occurs by loss of a proton to give the guanyl radical G(-H)•.
Since early works, it has been postulated that in DNA the proton is not directly lost from G•+ to the aqueous phase but remains within the hydrogen bonded G•+:::C pair located towards the cytosine N3 atom. The proposed mechanism consists of path 1 in Figure 3, wherethe population of positive charge in [G•+:::C D G(N1-H)•:::CH+] is estimate to be ~1:3 in favor of cytosine, based on pKa values, and in competition with the hydrolysis that give the 8-oxoG:::C lesion as stable product [14]. Tailored mechanistic studies were provided using1e– oxidation of ds-ODNs containing G:::C pairs followed up by various spectroscopic techniques. Using SO4•– as oxidant in pulse radiolysis studies, the decay of transient related to [G•+:::C D G(N1-H)•:::CH+] is associated with the release of the proton into solution. The rate constant for deprotonation was dependent on the ds-ODN sequence, especially on the sequence of bases adjacent to the guanine base, varying in the range 0.3–2 x 107 s–1 [15].
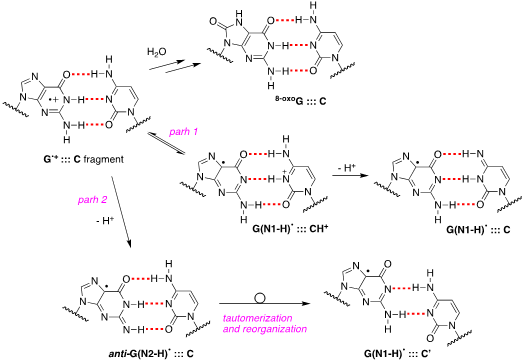
Figure 3. Proposed pathways of G•+:::C deprotonation in ds-ODNs (path 1 and path 2) and its reaction with water to give 8-oxoG:::C.
Figure 3 also shows path 2 where the protons can escape directly into the bulk solution forming anti-G(N2-H)•. The presence of anti-G(N2–H)•:::C in a ds-ODN is shown to be easily distinguished from the other prototropic forms, owing to its readily observable nitrogen hyperfine coupling by electron spin resonance (ESR) [16]. Indeed, the 1e– oxidation of d[TGCGCGCA]2 has been studied in details by ESR and UV-visible spectral analysis: at pH ≥7, the initial site of deprotonation is found to be at N1 forming G(N1–H)•:::C at 155 K and upon annealing to 175 K, the site of deprotonation to the solvent shifts to an equilibrium mixture of G(N1–H)•:::C and anti-G(N2–H)•:::C. This ESR identification is supported by the visible absorption at 630 nm which is characteristic for G(N2–H)• radicals.
The 1e– oxidation of ODNs by SO4•– for the quantification of 8-oxo-G lesion as final product was studied for comparison in ss-ODNs and ds-ODN [17]. The yield of 8-oxo-G in ds-ODN is ~7 times higher than in ss-ODNs indicating that the secondary structure of ds-ODN is a crucial factor that enhances the formation of 8-oxo-G lesions from G(N1-H)• radicals. The transient species observed by laser flash photolysis after the complete decay of SO4•– (5 μs) for both ds-ODN and ss-ODN were very similar and assigned to G(N1-H)•. However, the kinetics of G(N1-H)• decay were quite different: in ds-ODN, the decay is biphasic with one component decaying with a lifetime of ∼2.2 ms and the other one with a lifetime of ∼0.18 s, whereas in ss-ODN the decay is monophasic with a ∼ 0.28 s lifetime. The ms decay component in ds-ODN is correlated with the enhancement of 8-oxo-G yields which are ∼7 times greater than in ss-ODN. In ds-ODN, the authors proposed that the equilibrium [G•+:::C D G(N1-H)•:::CH+] allows for the hydration followed by formation of 8-oxoG:::C. By contrast, in ss-ODN, deprotonation of G•+ and the irreversible escape of the proton into the aqueous phase compete more effectively with the hydration mechanism, thus diminishing the yield of 8-oxo-G. In order to accommodate the fast deprotonation, they invoked path 2 in Figure 3 followed by a tautomerization of G(N2-H)• to G(N1-H)• coupled with base displacement and reorganization of the hydrogen-bonding network [17].
It was demonstrated that absorption of a single low-energy UV photon by ds-ODNs or ct-DNA at 265 nm generates guanyl radicals, with appreciable quantum yield (>10–3). The transient species detected after 3 μs are identified as guanyl radicals G(N1-H)•, with a half-life decay of 2.5 ms. The experimental results corroborated by theoretical studies suggest nonvertical processes, associated with the relaxation of electronic excited states [18].
It is well-known that some ODNs are arranged in G-quadruplexes. The structure of G-quadruplex consists of stacked G-quartets where each G-quartet is a planar array of four Hoogsteen-bonded guanines stabilized by metal cations. The existence of G•+ in G-quadruplex and their deprotonation from the NH2 moiety of guanines have been reported (Figure 4A) [19]. The three G-quadruplexes buffered using Na+ or K+, i.e., Tel22/Na+, TG4T/K+ and G4T4G4/Na+ that adopt antiparallel orientations are used (see Figure 12B). The one-electron oxidation was carried out by SO4•– using laser flash photolysis for the transient detection. The deprotonation rate of G•+ within G-quadruplexes (k = ~2 x 105 s–1) resulted 1−2 orders of magnitude slower compared to G•+ from ds-ODNs with formation of G(N2-H)•radical in anti-conformation (Figure 12A) [19].
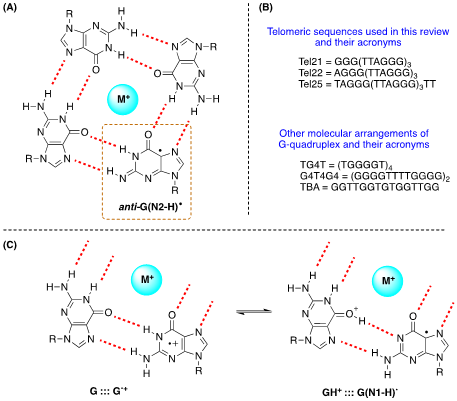
Figure 4. (A) Drawing of G-quartet containing the guanyl radical anti-G(N2-H)•; (B) The ss-ODNs sequences used in G-quadruplex structures; (C) Fragments of G-quartet; the proposed equilibrium in one-electron oxidized G quartet.
UV(266 nm)-induced one-photon ionization of G-quadruplexes and transient detection by ns time-resolved spectroscopy were also reported [20]. The fate of guanine radicals in the telomeric sequence Tel21/Na+ is followed over 5 orders of magnitude of time: (i) up to 30ns 100% G•+, (ii) in 3μs 50% G•+ and 50% anti-G(N2-H)• that become 35% G•+ and 50% G(N2-H)• in 20μs; (iii) in 5ms 50% G(N1-H)•that reaches 6% in 180ms. The two main features are a long-lived G•+ respect to ds-ODNs due to the secondary structure and the detection of the two guanyl radicals, including the tautomerization anti-G(N2-H)• → G(N1-H)• which takes place on the ms time scale. The TD-DFT calculations indicated that anti-G(N2-H)• is 12.1 kJ mol–1 more stable than the G(N1-H)• in simulated environment of Tel21/Na+ [20]. It is also worth mentioning the formation of 8-oxo-G in the comparative study on Tel21/Na+ and TG4T/Na+, where for the former the yield of 8-oxo-G is much higher than the latter [20]. Since the fate of G•+ splits between hydration vs. deprotonation, the authors suggested that the G•+ population survives on the ms time-scale in Tel21/Na+, whereas the deprotonation is much faster in TG4T/Na+, thus rendering the reaction path leading to 8-oxod-G less probable.
Other authors also investigated the 1e– oxidation of Tel25 by SO4•–, buffered using Na+ or K+, by laser flash photolysis for transient detection and analyzing the end-product 8-oxo-G [21]. In neutral aqueous solutions (pH 7.0), the observed transient is G(N1-H)• after the complete decay of SO4•– (~10 μs after the actinic laser flash). In both systems, the G(-H)• decay is biphasic with one component decaying with a lifetime of ~0.1 ms, and the other one with a lifetime of 20–30 ms. The fast decay component (~0.1 ms) in G-quadruplexes is correlated with the formation of 8-oxo-G lesions. The authors proposed that in G-quadruplexes, G(-H)• radicals retain radical cation character by sharing the N1-proton with the O6-atom of G in the [G•+:::G] Hoogsteen base pair (Figure 12C); this equilibrium [G:::G•+ D GH+:::G(N1-H)•] leads to the hydration of G•+ within the millisecond time domain, and is followed by the formation of the 8-oxo-G lesions [21].
In summary, the formation of G•+ in genetic materials and the mode of their disappearance depend on the secondary structure (ds-ODNs and G-quadruplex). This subject is still an active research topic although several articles and reviews addressed it in the last decade. Structural information and kinetics of the transient have been gained from time-resolved spectroscopies combined with theoretical calculations. Various mechanistic proposal are needed to be confirmed, since several questions remain open in both ds-ODNs and G-quadruplex systems: e.g., what is the mechanism of tautomerization anti-G(N2-H)• → G(N1-H)• in G-quadruplex, or what are the forces that drive the various deprotonation steps in G•+:::C of ds-ODNs, or what are the end-products of the two guanyl radicals and their relationship with 8-oxo-G lesion in G-quadruplex.





