B lymphocytes (B cells) are a key component of the human immune system. They can be isolated from peripheral blood or tonsils and be expanded in culture after activation by mitogenic agents. Here, it is demonstrated that a well-defined 24-armed poly(2-dimethylamino) ethyl methacrylate (PDMAEMA, 755 kDa) nano-star is able to reproducibly elicit high transgene expression (40%) at sufficient residual viability (69%) in primary human B cells derived from tonsillar tissue.
1. Introduction
B lymphocytes (B cells) are a key component of the human immune system. They can be isolated from peripheral blood or tonsils and be expanded in culture after activation by mitogenic agents
[1][2]. In recent years, the interest for genetic modification of human B cells has been growing, since B cells, in particular, those of the plasma cell subtype, are long-lived and have high protein production potential
[3]. Recombinant plasma B cells could thus, in future, be harnessed as “living drug delivery devices” for in vivo applications
[4][5][6][7][8][9]. Such an approach would improve current therapies for autoimmune diseases
[10], where repeated injections of neutralizing mAb are otherwise necessary to maintain a therapeutic titer. B cell engineering could also provide methods for vaccination against viruses such as HIV
[11][12][13] or lead to alternative therapies for the neutralization of overexpressed cytokines in chronic inflammatory diseases such as rheumatoid arthritis
[8]. In the treatment of genetic disorders, such as hemophilia B, engineered B cells could replace the costly periodic enzyme replacement therapies
[4][14].
Finally, several recent publications show that murine and human B cells can be efficiently edited by the clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) system (CRISPR/Cas9)
[4][12][13][14][15][16]. The examples range from the replacement of the endogenous antibodies by antibodies targeting viruses
[12][17] to the creation of chimeric B cell receptors presenting synthetic antigens
[8][12][13][18]. However, in order to use CRISPR-Cas9 in B cells, it is necessary to first deliver DNA encoding for the nuclease Cas9 and the synthetic guide RNA to the cells, where they are not naturally present
[19][20]. So far, in vitro delivery strategies for CRISPR/Cas9 into human B cells rely on viral vectors (mostly adenovirus, adeno-associated virus, lentivirus), which have high transfection efficiencies but are associated with a limited carrier capacity, immunogenicity, and the risk of insertional mutagenesis (for a review see
[14][20][21]). Non-viral gene delivery systems have been suggested as alternatives but, in the case of B cells, are severely limited by low transfection efficiency and high cytotoxicity
[20][21].
Better results were achieved with physical methods such as electroporation. Li et al., 2006, reached a transfection efficiency of 65% for a pDNA encoding for CD154 cDNA in chronic lymphocytic leukemia B cells (B-CCL), albeit with survival rates of only 30% 24 h post-transfection
[22]. Optimizing the salt concentration and the amount of pDNA may improve the efficacy of electroporation in peripheral primary B cells since, e.g., Canoy et al. reached 80% transfected cells at 55% viability
[23]. Moghimi et al. showed that Nucleofection, a specialized form of electroporation, may yield up to 65% transfected cells with 70% viability, whereas a liposome-based vector tested in parallel (i.e., Lipofectamine) never achieved more than 5% of transfected B cells
[24]. Such reports demonstrate that electroporation is at present the method of choice for non-viral gene delivery into B cells. However, as taken from these reports, electroporation requires large numbers of cells (up to 5 × 10
7 cells per electroporation mixture), which might not always be extractable from primary tissue material of a single donor. In addition, extensive amounts of pDNA are also needed (up to 440 µg pDNA per electroporation mixture), yet the production of high-quality pDNA is time-intensive and costly. In consequence, there is still a serious need for alternative non-viral procedures for gene delivery into primary B cells.
Cationic polymers represent a popular group of non-viral transfection agents for mammalian cell lines but are usually less capable of transfecting primary (immune) cells
[25][26]. An early report showed that low levels of transfection can be reached in primary murine splenic B lymphoblasts (i.e., LPS-activated B cells) using diethylaminoethyl (DEAE)-dextran
[27]. To the best of our knowledge, l-PEI and poly-2-(dimethylamino)ethyl methacrylate (PDMAEMA), i.e., two current standard polycations for the transfection of mammalian cell lines
[28][29][30], have never been proposed in the pertinent literature for the transfection of B cells with pDNA. In this context, PDMAEMA is a particularly interesting base structure for the design of improved non-viral agents because its chemistry allows precise control over molecular weight and structure during synthesis via atom transfer radical polymerization (ATRP)
[31]. In the past, we have proposed non-linear PDMAEMA nano-structures (stars, grafts) as efficient agents for nucleic acids delivery
[32][33][34][35] to “hard-to-transfect” cells, including primary human T lymphocytes
[36][37]. Recently, we showed that the superior transfection capability of nano-stars relies on the destabilization of plasma and nuclear membranes, which presumably leads to transient pore formation
[38], i.e., follows a transfection mechanism akin to electroporation.
2. Comparison of PDMAEMA-Nano-Stars to l-PEI for the Transfection of Primary Human B Lymphocytes
First, a standard approach to the development of a transfection protocol was used for both l-PEI (25 kDa), as the current gold standard in polycationic transfection agents, and the PDMAEMA based nano-stars. In this approach, the amount of pDNA was kept constant (3 µg), and the N/P ratio was adjusted by varying the amount of polycation used for polyplex formation. To get enough cells for parallel analysis of several transfection conditions (e.g., the N/P ratios), it was first necessary to expand the cells in growth medium for several days. To this end, the cells were cultivated with mitogenic factors (i.e., CD40L, IL-4, IL-21). Usually, four to six days were necessary to produce the required number of cells. Moreover, active division, which is accompanied by a temporal disassembly of the nuclear membrane, is generally accepted as a facilitator of transfection
[39].
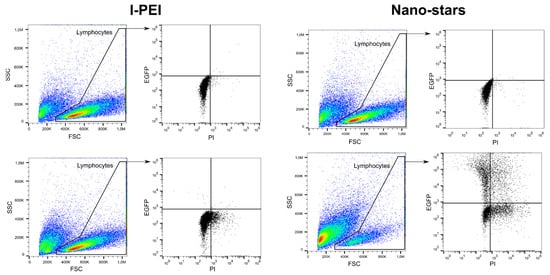
In this set of experiments, cells were transfected after four days of culture and incubated for four hours with the polyplexes in 6-well plates (transfection volume: 2 mL). The transfection outcomes (TE, survival) were analyzed 24 and 48 h post-transfection (Table 1) by flow cytometry (“Lymphocytes” gate). The gating strategy used in this process is shown in Figure 1.
Figure 1. Representative gating strategy for flow cytometry analysis. Top: mock transfection, bottom: Cells transfected with l-PEI (left) and nano-stars (right).
Table 1. Transfection of human primary B cells with PDMAEMA-nano-stars and l-PEI using the standard transfection method.
TE 1
(%) |
MFI 2
(a.u.) |
Viability
(%) |
| N/P Ratio |
Nano-Stars
24 h/48 h |
l-PEI
24 h/48 h |
Nano-Stars
24 h/48 h |
l-PEI
24 h/48 h |
Nano-Stars
24 h/48 h |
l-PEI
24 h/48 h |
| 3 |
4.9/2.4 |
3.7 /0.1 |
750/1085 |
495/715 |
66.3/48.8 |
90.9/95.9 |
| 5 |
9.3/6.5 |
1.8 /0.2 |
1289/2393 |
483/732 |
62.1/58.3 |
95.8/88.1 |
| 7.5 |
7.5/7.2 |
5.0 /0.3 |
1719/2099 |
500/732 |
60.9/54.3 |
77.7/83.9 |
| 12.5 |
24.3/8.1 |
1.9 /0.2 |
947/1311 |
496/784 |
46.6/44.9 |
80.5/90.5 |
| 15 |
15.1/6.2 |
2.7 /0.3 |
997/1096 |
503/778 |
52.9/31.2 |
75.1/93.8 |
| 20 |
20.5/4.1 |
4.1 /0.1 |
944/1103 |
500/730 |
55.5/42.3 |
80.7/87.3 |
1: TE, transfection efficiency. 2: MFI, median fluorescence intensity. Transfection in 6-well plates at day 4 post-thawing, pDNA: 3 µg, N/P ratio adjusted by varying the amount of polycation. Cell number for transfection: 2 × 105 cells. Contact time: 4 h. Transfection volume: 2.0 mL, n = 1. Cell viability on the day of transfection: >80%. a.u.: arbitrary units.
No replicates were tested since the number of primary cells from a given donor is, regardless of the expansion step, limited, while cells from different donors must be expected to vary in their experimental response
[40]. We limited our testing to N/P ratios corresponding to polymer concentrations ≤40 µg mL
−1 for nano-stars and ≤4 µg mL
−1 for l-PEI to avoid possible cytotoxic effects.
For the nano-stars, increasing the N/P ratio led to an increase in the TE with a concomitant decrease in viability. An N/P ratio of 20 allowed reaching a TE of ca. 20% in cultures 24 h post-transfection; however, only half of the cells survived. In terms of expression level, an N/P ratio of 7.5 seems to lead to the highest GFP production. In the case of l-PEI, the TE was always in the single-digit range, while the survival rate was around 80%. These results follow the general observation that high transfection efficiency is usually linked to greater cytotoxicity (as reviewed by Zhang et al., 2017)
[41]. Interestingly, whereas the TE decreased rapidly for both transfection agents with the cultivation time post-transfection, reaching values <1% for l-PEI and <10% for the nano-stars after 48 h post-transfection, the expression level, indicated by the median fluorescence intensity (MFI), increased in all cases. This result indicates that the remaining transfected living cells are transcriptionally active.
In our experimental setup, transfection was supposed to be transient, i.e., no active integration into the genome was intended. However, such a rapid decrease in TE was not expected and is usually not observed in cell lines transfected according to similar protocols, where GFP accumulation can typically be observed for at least 72 h
[42]. An explanation for this behavior can only be speculated upon. In the past, Seiffert et al. reported that circular pDNA induces apoptosis in nucleofected primary B cells
[43]. It has also been reported that exposure of cells to apoptotic stimuli induces a rapid loss of cell volume, the so-called apoptotic volume decrease
[44]. Since we restricted our analysis to the lymphocyte population identified by scattering properties, a significant decrease in the cell volume during the incubation post-transfection would lead to a shift of these cells outside of the “Lymphocytes” gate (i.e., smaller forward scatter) and
inter alia decrease the TE evaluated in this gate.
The better survival of the cells in case of transfection with l-PEI may also be ascribed to the lower polymer densities (6.0 to 39.0 µg per 10
6 cells for l-PEI, 22.0 to 144.0 µg per 10
6 cells for the nano-stars) and polymer concentrations (0.6 to 4.0 µg mL
−1 for l-PEI, 2.0 to 14.0 µg mL
−1 for the nano-stars) required to reach the indicated N/P ratios (see
Table S1 for details). However, for both polycations, the polymer concentration at the highest N/P ratio was still below the LD
50 values recorded for free l-PEI (12 µg mL
−1) and nano-stars (39 µg mL
−1) in L929 cells (MTT assay) by our group
[45]. Previously, we have shown that human primary T cells have a two-fold higher sensitivity to these polycations than the L929 cells and some similarity can be presumed for primary B cells
[37]. Still, none of the polymers were expected to be toxic in the concentration range tested. Since cells generally tend to tolerate nano-stars better than l-PEI, the negative effects on viability observed here for the nano-stars may be associated with cellular “disorders” post-engulfment of the polyplexes, which built up during the post-transfection period. This is corroborated by data recently obtained by us for Jurkat cells
[38]. A similar effect is not expected for l-PEI, which has a much lower tendency to enter the cells (low TE) and thus is removed during washing.
3. Influence of the Cell Number and the Polymer Density (Amount per Cell)
In the past, we were able to show that changing the geometry of the transfection vessel from plate to tube allows reducing the reaction volume, thereby
inter alia intensifying the interactions between cells and polyplexes, while concomitantly accelerating the transfection kinetics. As a result, the tube transfection protocol highly improved the transfection outcomes of some “hard-to-transfect” cells in terms of TE and viability post-transfection
[36]. Here, we hypothesized that a similar benefit may be possible in the case of the primary B cells. For fine-tuning the conditions, we investigated the influence of the cell number, but also the polymer density (µg per 10
6 cells) using quantities identified as most efficient for gene delivery into human T cells as starting conditions
[36].
For the investigation of the cell number, 2, 3, or 5 × 10
5, cells were incubated for 90 min with polyplexes formed at an N/P ratio of 10, i.e., a ratio sufficient for charge neutralization of the pDNA when using nano-stars
[36]. Since preliminary screening experiments had shown that polymer densities ≤ 10 µg polymer per 10
6 cells (≤4 µg polymer per mL) led to TE < 15%, experiments were carried out at polymer densities of 15 to 25 µg polymer per 10
6 cells (6 to 25 µg polymer mL
−1), the N/P ratio of 10 was assured in each case via the added pDNA. Transfection efficiencies (TE) and the cell viabilities were analyzed 48 h post-transfection by flow cytometry (
Figure 2), as prescribed by Riedl et al. for primary T cells
[36]. If anything, a measurement after 48 h constitutes a “worst case scenario”, since in the experiments described above, the TE tended to decrease with post-transfection cultivation time.
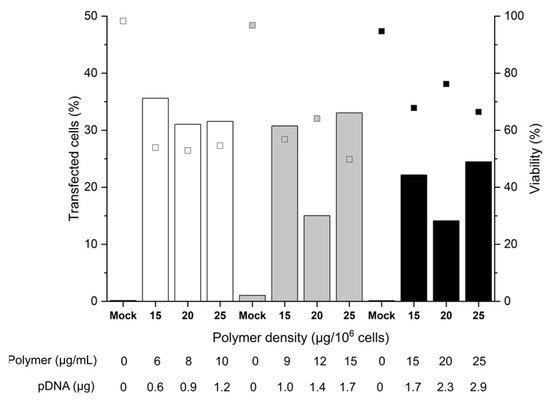
Figure 2. Influence of cell number and polymer density on transfection efficiency and viability. Transfection in tubes at day 6 post-thawing. Cell numbers during transfection were: 2 × 105 (white), 3 × 105 (grey), 5 × 105 (black) cells. pDNA corresponds to the amount of plasmid per experiment. Contact time: 90 min. N/P: 10, transfection volume: 0.5 mL. TE (bars) and viability (squares) measured 48 h post-transfection. “Mock”: cells subjected to mock transfection. n = 1. Cell viability on the day of transfection: 81%.
A maximum TE of 36% was achieved with 2 or 3 × 105 cells per tube, which was a considerable improvement over the plate protocol, where TEs generally were below 10% after 48 h. A further increase in the cell number to 5 × 105 cells per tube reduced the TE 1.8-fold. This could be related to insufficient mixing of cells and polyplexes due to the higher viscosity of the mixture. Strikingly, for 3 and 5 × 105 cells, at identical polymer concentration (15 µg mL−1) and pDNA amount (1.7 µg), a significant increase in TE/decrease of viability correlated with an increase in the polymer density during transfection. Hence, an increase in the number of polyplexes per cell (i.e., polyplexes dose) improves the TE to the detriment of cell viability.
On the other hand, if we assume the otherwise difficult to explain lower TEs measured with 20 µg polymer per 10
6 cells for 3 and 5 × 10
5 cells per tube to be due to experimental error, the polymer density has no major influence on the transfection outcomes for a given number of cells. In the past, we observed similar trends for the Jurkat cells
[36]. The fact that increasing the polymer density (i.e., the polyplex dose, but also the pDNA dose) for a given number of cells does not influence the overall TE, could be a first indication that some saturation of one or several rate-limiting steps of the transfection (cellular and/or nuclear uptake, decomplexation of polyplexes, or other) occurs. Hence, for a given polymer density, there is an upper limit of effectivity above which further raising of the polymer density has no beneficial effect and only reduces the cell survival. Furthermore, overloading the transcriptional machinery due to an excess of pDNA in the nucleus (i.e., a titration of the transcription factors) cannot be excluded. On the other hand, a decrease in the relative polyplex dose via increasing the cell number at a given amount of polyplexes negatively influences the TE. We can hypothesize that in that case, the average number of polyplexes per cell is reduced (in line with the observed trend towards improved viabilities), which leads to a reduced level of GFP expression. In general, cells expressing GFP can be readily detected by flow cytometry. However, a precise distinction of cells with low expression levels from the autofluorescence of non-transfected cells is difficult due to a distribution overlap. Hence, the observed TE might be underestimated.
The survival rates of the B cells showed some variability (between 50 to 70%) but were tendentially better than in the plate protocol, despite the improved TEs. The mock-transfected cells exhibited viabilities > 90%, indicating that the transfection procedure per se had no negative effect on B cell viability within the 48-h time frame of observation. Remarkably, within the range tested, the polymer concentration adjusted during transfection did not itself influence the cell viability. This first set of data indicated that 2 × 105 cells per tube and polymer densities between 15 and 25 µg mL−1 are well-suited conditions that can be taken as a basis for further optimization.
4. Influence of the Polyplex Exposure and the Recovery Times in B Cell Transfection
Considering that the nano-stars induce a transient poration of the plasma membrane
[38], the prolonged 90 min-exposure to the polyplexes as prescribed in the standard protocol might have had negative consequences in sensitive primary cells, which have a less robust plasma membrane repair machinery than, e.g., cancer cells
[46]. To test the hypothesis that a reduction in the contact time may be of benefit, 2 × 10
5 cells were incubated for 10 to 90 min with polyplexes (N/P 10, corresponding to 15 to 30 µg polymer per 10
6 cells or 6 to 12 µg polymer mL
−1). The limited cell number recovered from individual batches after freezing/thawing obliged us to do the testing on two batches of thawed cells (same donor) to cover the entire time range we wanted to study (group A: 10 to 30 min, group B: 30 to 90 min). Experiments were started as soon as sufficient biomass was available in the sum of the two batches, i.e., on day 4 post-thawing. Transfection efficiencies and cell viabilities were again measured 48 h post-transfection by flow cytometry (
Figure 3).
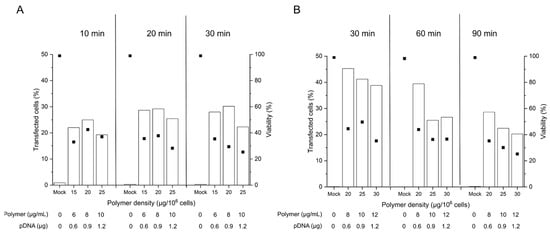
Figure 3. Influence of the polyplex contact time on transfection efficiency and viability. Cell number during transfection: 2 × 105 cells (day 4 post-thawing), tube transfection protocol. pDNA corresponds to the amount of plasmid per tube. Contact time: as indicated, N/P: 10, transfection volume: 0.5 mL. TE (bars) and viability (squares) were measured 48 h post-transfection. Mock: cells subjected to mock transfection. n = 1. Cell viability on the day of transfection: 80%, group A (A) and 93%, group B (B).
Even though issued from the same donor tissue, the obtained B cell pools showed differences in the overall viability on the day of transfection, which was 80% in group A and 93 % in group B. As reported in the past, B cells show some susceptibility to freezing damage
[47], which may influence their fitness after thawing. In consequence, we did our best to standardize the procedures, however with mitigated success.
The TEs obtained in this experiment for the 90 min incubation were in the range shown before for cells were transfected 6 days post-thawing (Figure 2), which suggests that a reduction in the cultivation time before transfection has little effect on the transfection outcome in terms of TE. Lowering the polyplex exposure time from 90 to 60 and 30 min has a beneficial effect on the TE as well as on the viability of the cells, while yet lower exposure times show no additional benefit. A 30 min exposure to the polyplexes may thus be considered an optimum contact time for the transfection of primary B cells according to the tube protocol. However, under experimentally similar conditions, group A exhibited lower TEs and viabilities than group B for a given polymer density at 30 min exposure. This may well be due to the lower fitness (viability) of the cells from group A on the day of transfection. In cell lines, a viability of >90% is typically prescribed in standard non-viral transfection protocols, a quality, which is not always achievable in primary cells.
To further investigate the extent of possible intra- and inter-experimental variations, cells recovered from five cryovials (same donor) were transfected after 4 days of cultivation in the presence of mitogens as follows: 2 × 105 cells were incubated for 30 min with polyplexes corresponding to 15 µg polymer per 106 cells (6 µg polymer mL−1, N/P ratio: 10). Transfection efficiencies and cell viabilities were measured 48 h post-transfection (Table 2). Whereas the intra-experimental variation (technical replicates) was low (3.6% for TE; 1.4% for viability), the inter-experimental variation (experimental replicates) was more pronounced (9.2% for TE; 11.6% for viability). It should be noted that in these experiments as well, higher viability on the day of transfection seemed to correlate with an improved transfection outcome.
Table 2. Influence of the batch variation on transfection efficiency and viability.
| Cryovial Nb. |
Viability Pre-Transfection
(%) |
Transfection Efficiency
(%) |
Viability Post-Transfection
(%) |
| I 1 |
|
22 |
33 |
| |
79.9 |
28.6 |
35.6 |
| |
|
28 |
35.4 |
| II |
90.4 |
41.3 |
54.1 |
| III |
84.4 |
16 |
46.2 |
| IV |
86.9 |
31.8 |
52.9 |
| V |
82.6 |
40.8 |
63.6 |
| Mean ± SD (%) |
83.4 ± 4.1 |
29.8 ± 9.2 |
45.8 ± 11.6 |
Transfection in tubes. Cell number during transfection: 2 × 105 cells (day 4 post-thawing). Polymer density: 15 µg per 106 cells, polymer concentration: 6 µg mL−1, pDNA: 0.6 µg per tube. Contact time: 30 min. N/P: 10, transfection volume: 0.5 mL. TE and viability were measured 48 h post-transfection. n = 1. 1: Technical replicates, meanTE: 26.2 ± 3.6 % and meanviability: 34.7 ± 1.4 %.
Finally, the transfection capability of l-PEI in the tube protocol was tested. In these experiments, we kept the experimental setup as similar as possible to the conditions used for nano-stars (Table 2) but extended the range of N/P ratios tested. After 48 h of cultivation post-transfection, the transfection efficiency was below 0.5% in all cases with viabilities ≥73%. These results underline that a switch to the tube protocol has no benefit in the transfection of B cells with l-PEI.
To fully investigate any effect of the post-thawing recovery time on the day of transfection, cells expanded from various cryovials from the same donor were transfected three to five days post-thawing. During this time interval, the cells were in the exponential phase (growth rate: 0.071 h−1). The TE and the cell viabilities were analyzed 24 to 48 h post-transfection in order to concomitantly follow the development of these two parameters with cultivation time, Table 3. Again, a pronounced heterogeneity was observed between the batches thawed, even though the cryovials were prepared from the same pool of cells isolated from one donor. Cell viability, in particular, varied from 65 to >90% after three to five days of cultivation post-thawing. To reduce at least the influence of the cell viability on the day of transfection, we only used the expanded cells if their viability was >80%.
Table 3. Transfection outcomes as a function of the pre- and post-transfection cultivation time.
| Cultivation Time |
|
|
|
| |
Pre-Transfection
(Days) |
Post-Transfection
(Hours) |
TE (%) |
MFI 2 (a.u.) |
Viability (%) |
| Tfd3 |
3 |
24 |
39.2 ± 0.9 |
34,537 ± 1532 |
61.8 ± 0.3 |
| (n = 3) |
|
| 48 |
28.2 ± 6.1 * |
32,220 ± 2695 |
53.0 ± 1.6 * |
| (n = 3) |
|
| Tfd4 1 |
4 |
24 |
37.4 ± 1.8 |
41,564 ± 11,966 |
69.3 ± 1.7 # |
| (n = 2) |
|
| 48 |
32.5 ± 3.9 |
35,204 ± 9644 |
54.2 ± 2.4 * |
| (n = 4) |
|
| Tfd5 |
5 |
24 |
n.a |
n.a |
n.a |
| 48 |
21.2 ± 5.8 § |
10,940 ± 759 |
38.3 ± 9.8 # |
| (n = 3) |
|
1: For analytical sake, some of the transfection data included in the calculation were identical to the one presented below in § 2.4. 2: MFI, median fluorescence intensity. 2 × 105 cells per samples; N/P 10; 15 µg polymer per 106 cells; 6 µg polymer per mL. Contact time: 30 min. Transfection in tubes, transfection volume: 0.5 mL. Cell viability on the day of transfection: >80%. Data represent mean values ± SD. n.a.: not available. Statistical significance between “day of cultivation pre-transfection” groups is indicated as # (p < 0.05). Statistical significance between Tfd5 and Tfd3 or Tfd4 is indicated as § (p < 0.05). Statistical significance between “24 h” and “48 h” groups is indicated as * (p < 0.05).
According to the results compiled in Table 3, the cells seemed to be most tolerant towards the transfection conditions on day 4, since the viability was highest. TEs were in that case similar to day 3, while TE levels of GFP expression and viability dropped for cells transfected on day 5. However, the trend towards a higher GFP expression when the cells were transfected at day 4 post-thawing was not statistically significant. As observed before for transfection, according to the plate protocol, both the TE and the viability of the cells tended to deteriorate 48 h post-transfection compared to the values recorded after 24 h. The difference was statistically relevant for cells transfected on day 3 of cultivation and may thus indeed present a general problem in B cell transfection.
 +1 point
+1 point