To maintain homeostasis, the lung ECM undergoes remodelling, a process whereby old or damaged ECM proteins undergo a series of proteolytic events and are replaced by newly synthesized proteins. Lung ECM remodeling is modulated by the synthesis rate of new ECM molecules and the surfeit of proteases released by specialized mesenchymal cells, predominantly fibroblasts and, to a lesser extent, airway smooth muscle, immune cells and epithelial cells of the lungs
[4][10]. The remodelling process is directed by the interaction between the different cell types, cytokines, growth factors and enzymes within the lung ECM. The levels and ratios of ECM proteins during the repair response must be maintained to avoid disrupting the lung ECM characteristics, including tensile strength and elastic recoil. Upon lung injury, activation of the coagulation process initiates damage control and a provisional matrix (fibronectin), which is then followed by the development of an acute inflammatory response that recruits polymorphonuclear leukocytes (PMNs) and macrophages to protect against pathogens and remove debris from dead and dying cells. These important early steps prepare the site for activation of local fibroblasts, migrating myofibroblasts and recruited fibrocytes that remodel collagenous and noncollagenous tissues into scar tissue. Following a single injury, inflammatory cells usually leave the site before the resident fibroblasts and migrating myofibroblasts, and fibrocytes are activated to drive the tissue repair process. Over time, the provisional ECM is degraded via cell-mediated or proteolytic pathways and replaced with a restorative ECM consisting mainly of collagen I and collagen III, before these cells then undergo apoptosis
[11]. In sharp contrast, repetitive injury is associated with the persistence of inflammatory immune cell infiltration during the remodelling of the ECM, which increases the potential for the development of an abnormal repair process creating further injury with abnormal scar formation, remodelling or permanent destruction of damaged tissue. Repeated lung injury and repair can trigger a vicious cycle of aberrant ECM protein deposition, accompanied by elevated ECM stiffness, further resulting in changes in cell phenotype and function, and this has a lasting effect on tissue function and ultimately disease progression
[12]. Although the processes governing the resolution of injury repair are regulated by several pathways, in chronic fibrotic lung diseases the processes are compromised, thus resulting in impaired fibroblast proliferation, apoptosis and aberrant ECM remodelling
[3][11]. Disruption of this balance changes the dynamics of the lung ECM with characteristics such as stiffness and elastance, as seen in asthma, chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF)
[13][14][15]. Compared to the normal lung ECM (0.5–5 kPa), studies have shown that a stiff ECM (15–100 kPA)
[16][17][18][19] can exacerbate TGF-β activation leading to enhanced pro-fibrotic signalling. Disruption of homeostasis within the lung ECM can also lead to the dysregulated synthesis of ECM proteins, such as fibrillar collagen I, by activated fibroblasts, leading to tissue fibrosis regardless of the inflammatory response
[20]. The interplay between several diverse factors (cell types, genes, cytokines, growth factors, enzymes and epigenetics) is essential for the normal repair and remodeling processes within the lung
[21]. Studies have shown that deviations in the posttranslational modification of ECM proteins, including enzymatic cross-linking, glycation and oxidation, impact the tensile strength, biomechanics and cell–ECM interactions and are contributors to lung diseases
[22][23]. As shown in
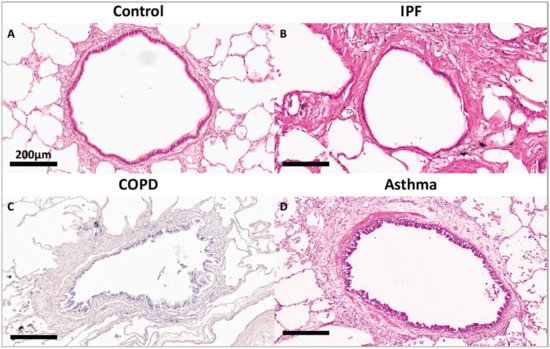
Figure 1, compared to a terminal bronchiole (last generation of conducting airways within the lung) from a donor control, there is extensive fibrosis surrounding the terminal bronchiole airway walls in the lungs from patients with IPF, COPD and asthma.
 +1 point
+1 point