1000/1000
Hot
Most Recent
 +1 point
+1 point
Mucopolysaccharidosis-plus syndrome (MPSPS) is a novel disease of impaired glycosaminoglycans (GAGs) metabolism without deficiency of known lysosomal enzymes. MPSPS, whose pathophysiology is not elucidated, is an autosomal recessive multisystem disorder caused by a specific mutation p.R498W in the VPS33A gene. Clinical features of MPSPS patients are similar to conventional mucopolysaccharidoses (MPS). In addition to typical symptoms for conventional MPS, MPSPS patients developed other features such as congenital heart defects, renal and hematopoietic disorders. Diagnosis generally requires evidence of clinical picture similar to MPS and molecular genetic testing. Disease is very severe, prognosis is unfavorable and most of patients died at age of 10–20 months. Currently there is no specific therapy for this disease and clinical management is limited to supportive and symptomatic treatment.
Mucopolysaccharidosis-plus syndrome (MPSPS) is a novel disease of impaired glycosaminoglycans (GAGs) metabolism without deficiency of known lysosomal enzymes. MPSPS, whose pathophysiology is not elucidated, is an autosomal recessive multisystem disorder caused by a specific mutation p.R498W in the VPS33A gene.
Diagnosis generally requires evidence of clinical picture and molecular genetic testing. The finding of elevated urinary GAGs is supportive. Diagnosis is based upon on identification of detailed patient and family history. Prenatal diagnosis can verify if a fetus is affected with MPSPS.
All MPSPS patients showed recurrent infections of the upper respiratory tract in infancy or early childhood. MPSPS is a multisystem disorder with severe phenotype. In addition to clinical phenotype of conventional MPS, MPSPS patients present with hematopoietic disorders, renal involvement and congenital heart defects. Individuals affected by MPSPS share similar symptoms that progress rapidly.
Most patients develop hematopoietic disorders including anemia, thrombocytopenia and leukocytopenia. An increase in serum IgM and decrease of IgG levels were found in MPSPS patients[1]. Bone marrow aspiration analysis showed hypoplastic bone marrow without any storage cells. Radiograph examinations showed signs of dysostosis multiplex: J-shaped sella turcica, bullet-shaped phalanges and the metacarpal pointing, metaphyseal widening of the long bones with cortical thinning, oar-shaped ribs, flared iliac wings and malalignment of vertebrae and vertebral dysplasia with round- and hooked-shape vertebral bodies (Figure 1). Magnetic resonance imaging (MRI) and computed tomography (CT) of the brain showed apparent delayed myelination in the peripheral white matter and calcification in the basal ganglia. Echocardiography usually identifies atrial septal defect, patent ductus arteriosus or hypertrophic cardiomyopathy. Retinal hypopigmentation was seen on the fundus photograph[2]. Electron microscopy of conjunctiva biopsy revealed early and late endosomes in endothelial cells, myelin figures and enlarged mitochondria. The majority of MPSPS patients had a nephrotic syndrome. Proteinuria were in the range from mild to severe. Patients have an increased excretion of uronic acid in urine. Renal ultrasonography shows nephromegaly and increased echogenity[2]. Renal biopsy showed segmental sclerosis, periglomerular fibrosis and inflammatory cell infiltration.
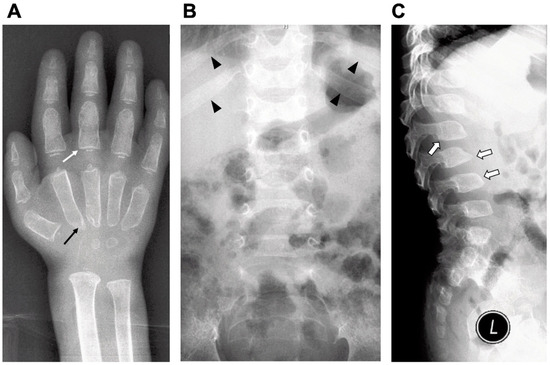
Figure 1. Radiographic findings of a MPSPS patient at age of one-year and nine-month. Characteristics of dysostosis multiplex: (A) bullet-shaped phalanges (white arrow) and the metacarpal pointing (black arrow); (B) widening of anterior ribs (oar-shaped ribs) (arrowhead) and (C) vertebral dysplasia with round- and hooked-shape vertebral bodies (open arrow).
Examination of urine can reveal elevated levels of GAGs, specifically heparan and dermatan sulfate[1][2][3]. Accumulation of heparan sulfate was also detected in patient’s plasma and skin fibroblasts. Moreover, the heparan sulfate level in plasma was significantly exceeded in comparison with conventional MPS[2]. Activities of lysosomal glycosidases involved in the degradation of GAGs are not decreased in plasma, lymphocytes and patient-derived fibroblasts[2][3]. Finding of MPS-like phenotype and excess secretion of urinary GAGs without deficiency in the activity of lysosomal enzymes is the basis for suspicion of MPSPS. However, these tests cannot be used to provide definitive diagnosis of MPSPS. Diagnosis should be confirmed by molecular DNA analysis.
MPSPS is caused by missense mutation p.R498W in the VPS33A gene and is inherited in an autosomal recessive pattern. This mutation is specific, and no other disease-causative mutation has been reported in the VPS33A gene. Parents of MPSPS patients are heterozygous carriers of p.R498W mutation. Heterozygous carriers for MPSPS mutation are generally disease free and do not develop symptoms of disease. MPSPS affects males and females in equal numbers. The frequency of the pathogenic allele was very low in diverse populations from different databases (Table 1). But among Yakut population we observed extremely high allele frequency.
Table 1. Allele frequency of c.1492C>T VPS33A (p.R498W, rs767748011).
|
Population (database) |
Allele Count |
Allele Numbers |
Allele Frequency |
|
|
ExAC database |
2 |
118,008 |
1:59,004 |
|
|
TOPMED database |
1 |
125,568 |
1:125,568 |
|
|
GnomAD database |
2 |
245,570 |
1:122,785 |
|
|
Ensembl database |
|
|
|
|
|
|
African |
0 |
15,260 |
0:15,260 |
|
|
European (Finnish) |
0 |
22,218 |
0:22,218 |
|
|
European (Non-Finnish) |
2 |
111,244 |
1:55,622 |
|
|
Ashkenazi Jewish |
0 |
9828 |
0:9828 |
|
|
East Asian |
0 |
17,236 |
0:17,236 |
|
|
South Asian |
0 |
30,770 |
0:30,770 |
|
|
Latino |
0 |
33,544 |
0:33,544 |
|
Yakut (our study) |
5 |
404 |
1:81 |
|
Clinical features of MPSPS patients are similar to conventional MPS. In addition to other forms of MPS, the differential diagnosis should also include LSD, such as mucolipidosis types II and III, Niemann-Pick disease, Gaucher's disease, fucosidosis, mannosidosis, sialidosis and multiple sulfatase deficiency. Clinical and biochemical studies distinguish these conditions. Disease is very severe, prognosis is unfavorable. Most of patients died of cardiorespiratory failure at the age of 10–20 months (Figure 2).
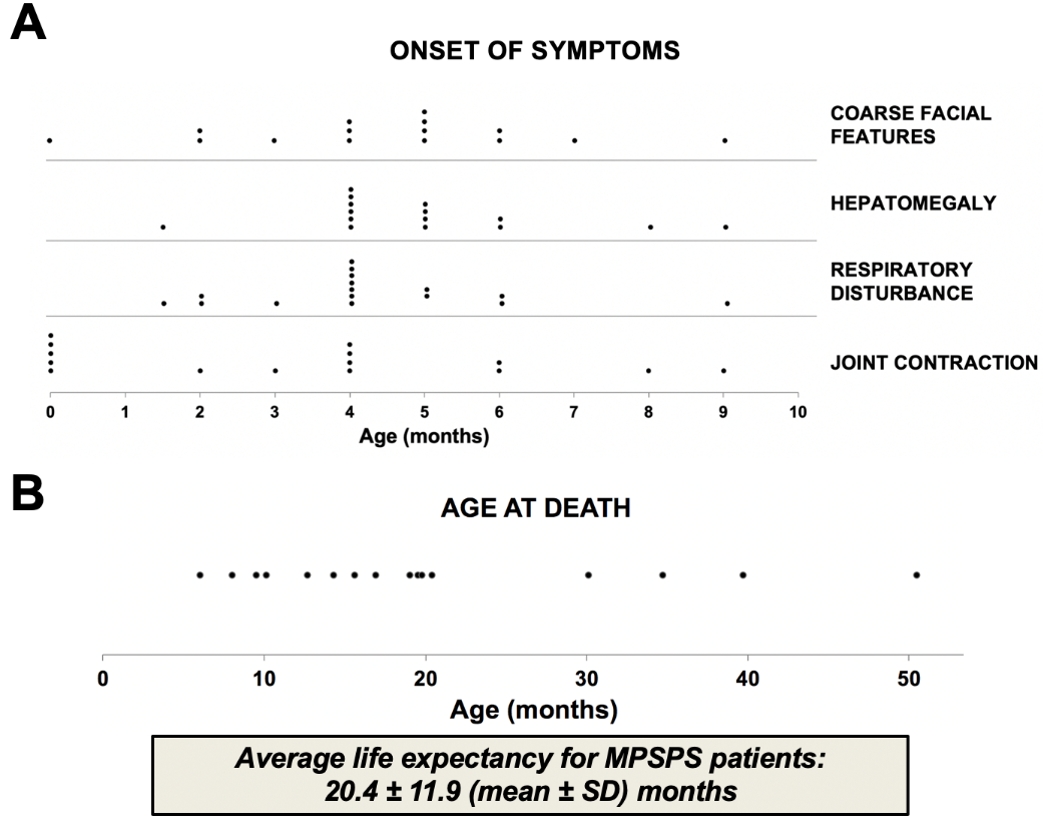
Figure 2. Onset of clinical features and life expectancy of MPSPS patients. Data from sixteen MPSPS patients from Yakutia (Russia). (A) Symptoms of disease usually occur between the ages of 2–6 months. (B) Most of patients died of cardiorespiratory failure at age of 10–20 months. Each dot indicates one MPSPS patient.





