1000/1000
Hot
Most Recent
 +1 point
+1 point
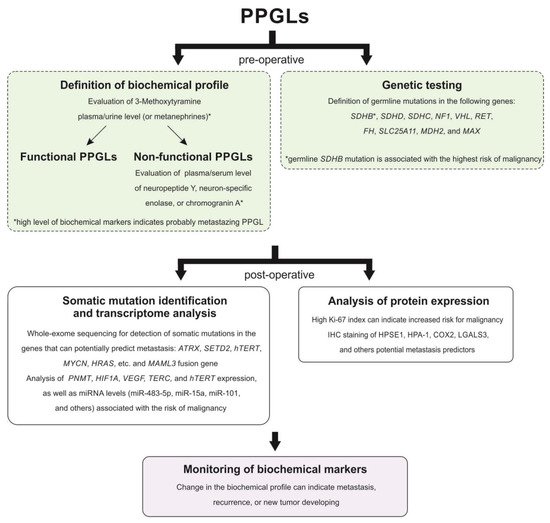
Paragangliomas and pheochromocytomas (PPGLs) are rare neuroendocrine tumors formed from paraganglionic tissue. Since 2017, PPGLs have been classified as tumors with variable potential to metastasize. Metastasizing PPGLs are usually difficult to diagnose and require evidence of regional or distant metastasis. Data on diagnostic and prognostic molecular markers for PPGL malignancy are limited, and many of the proposed factors remain controversial. There is a significant gap in the understanding of tumor pathogenesis, as well as the treatment and management of patients with PPGLs. This entry summarized the current findings on the potential markers for distinguishing between metastasizing and benign tumors, as well as on the prediction of aggressive behavior of PPGLs, especially of those localized in the head and neck region.
| Parameter | HNPGLs | PHEO | Other Extra-Adrenal PGLs | ||
|---|---|---|---|---|---|
| CPGL | MEPGL | VPGL | |||
| Mean age at diagnosis | 40–50 * [7][8] | 55 [3] | 41–47 [3] | 40–50 [9] | 40–50 [9] |
| Female/male ratio | 2:1–8:1 ** [10][11] | 3:1–9:1 [12] | 2:1–8:1 [12] | 1:1 [9] | 1:1 [9] |
| Multifocal cases, % | 10–25 [4] | 10–50 [13] | 10 *** [14] | 8 [15] | 33 [15] |
| Metastatic cases, % | 4–6 [12] | 2 [12] | 16–19 [10][12] | 10 [9] | 2.5–50 [9] |
Among the main genetic features associated with a high risk of the development of metastasizing PPGLs is a germline mutation in the SDHB gene. Testing for the germline SDHB mutation in patients with PPGLs is recommended by Clinical Practice Guidelines [26][27]. According to a systematic review and a meta-analysis study, the pooled incidence risk of metastasizing PPGLs for the SDHB mutation carriers was 17% while the prevalence ranged from 13% to 23% [51]. Among the patients with HNPGLs, the reported incidence of metastasis reaches 83% in the groups of SDHB mutation carriers.
The germline mutation in the SDHD gene was also reported in metastasizing PPGLs; however, the risk of metastasis development in SDHD mutation carriers is significantly lower than in those with an SDHB mutation [52]. SDHD mutations are more frequently associated with HNPGLs and multiple tumors [53][54][55]. The pooled risk of incidence and prevalence of metastasizing PPGLs for SDHD mutation carriers was estimated as 8% and 3%, respectively [51]. The incidence risk of malignancy for patients with SDHD-mutated HNPGLs reaches 22.7%. The highest incidence risk of malignancy (100%, 4/4) was observed among patients with HNPGLs from the Dutch population; at the same time, no variants were found in the SDHB gene [56]. All these patients carried a founder mutation in the SDHD gene. Thus, this higher association of the SDHD mutation with malignancy compared with SDHB, which was found in most studies, can be explained by characteristics of the Dutch population.
Several studies reported germline mutations in other susceptibility genes for PPGLs, such as FH [57], SLC25A11 [58], and MDH2 [59], which were associated with aggressive tumor behavior. These genes are classified as cluster 1 TCA cycle-related associated with the pseudohypoxia subtype of PPGLs [60]. Moreover, tumors with mutations in these genes were clustered together with SDHx-mutated tumors demonstrating similar hypermethylation profiles [61][58][59][62]. This phenotype seems to be involved in tumor progression and mutations in the FH, SLC25A11, and MDH2 genes along with SDHB and SDHD mutations can be considered to be a risk factor for PPGL malignancy. However, mutation frequency in these genes is rare and accounts for less than 1% [57][58][63]. Notably, alterations in FH and SLC25A11 were found in HNPGLs but in non-metastasizing tumors [57][58][63].
ATRX is a frequent somatically mutated gene in PPGLs. The most frequent ATRX alterations have been observed in SDHx-mutated tumors, including metastasizing PPGLs [64]. Moreover, several studies showed that the somatic ATRX variant occurring with the SDHB mutation and/or TERT overexpression was an indicative marker of metastasizing tumors [65][66]. An important role of ATRX and telomere maintenance mechanisms during tumor progression was also confirmed by the presence of alternative lengthening of telomeres (ALT) in ATRX-mutated metastasizing PPGLs [67].
The Ki-67 protein is another important biomarker of tumor progression used in grading systems and prognosis prediction for several types of cancer [68]. It is also included in the pathological grading system GAPP for the estimation of metastatic potential in PPGLs. PPGLs usually have low proliferation activity with the Ki-67 score varying from 0% to 2%. However, elevated proliferation activity (over 2%) was observed in metastasizing PHEOs and PGLs [22][23][69]. Moreover, a series of studies have reported metastasizing PPGLs with the Ki-67 index of more than 4% [21][70]. Nevertheless, metastatic tumors can also have proliferation activity up to 2%, indicating the high specificity but low sensitivity of the method [71][72]. Recent research by Guo et al. showed an association between the Ki-67 index and the programmed death ligand 1 (PD-L1) expression in PPGLs [73]. Although PD-L1 expression was not significantly correlated with the presence of distant metastases, PD-L1 positivity in tumor cells with high Ki-67 may indicate that cells acquire the ability to escape the immune system, contributing to tumor growth, invasion, and metastasis [73][74]. The association of tumor progression with immune evasion in PPGLs was confirmed by the fact that almost half of the metastasizing PPGLs expressed PD-L1 or PD-L2 [74]. Additionally, the TCGA project study found a positive correlation of the Ki-67 index with metastasizing PPGLs and its highest expression in MAML3 fusion-positive tumors related to the Wnt signaling cluster. This indicates that the activation of the Wnt signaling pathway can promote tumor cell proliferation and progression of paragangliomas [75].
| Potential Marker | Characteristics Associated with Malignancy |
|---|---|
| Histopathological markers | |
| Grading system for adrenal pheochromocytoma and paraganglioma (GAPP) | Well-differentiated and moderately differentiated tumors |
| Tumor size and weight | On average, larger than 10 cm and more than 500 g |
| Adrenal gland scaled score (PASS) | ≥4 |
| Ki-67 proliferation index | >2% |
| Sustentacular cells | Cell density depletion or absent |
| Galectin-3 (LGALS3) | Increased expression detected using IHC staining |
| Succinate dehydrogenase complex subunit B (SDHB) | Negative or weak diffuse IHC staining |
| Heparanase-1 (HPSE1) | Positive IHC staining |
| Cyclooxygenase-2 (COX2) | |
| Genetic markers | |
| Succinate dehydrogenase complex subunit B (SDHB) | Germline mutation |
| Succinate dehydrogenase complex subunit D (SDHD) | |
| Fumarate hydratase (FH) | |
| Solute carrier family 25 member 11 (SLC25A11) | |
| Malate dehydrogenase 2 (MDH2) | |
| ATRX chromatin remodeler (ATRX) | Somatic mutation |
| Histone-lysine N-methyltransferase SETD2 (SETD2) | |
| Telomerase reverse transcriptase (hTERT) | |
| Mastermind-like transcriptional coactivator 3 (MAML3) | Fusion gene |
| CpG island methylator phenotype (CIMP) | High CIMP |
| MicroRNA miR-15a | Downregulation |
| Phenylethanolamine N-methyltransferase (PNMT) | |
| MicroRNA miR-483-5p | Overexpression |
| MicroRNA miR-101 | |
| MicroRNA miR-210 | |
| MicroRNA miR-21-3p | |
| MicroRNA miR-183-5p | |
| Telomerase reverse transcriptase (hTERT) | |
| Biochemical markers | |
| Normetanephrine and 3-methoxytyramine | Increased plasma or urine level |
| Neuron-specific enolase (NSE) | Increased serum level |





