1000/1000
Hot
Most Recent
 +1 point
+1 point
Melanomas are diagnosed at various stages of the disease and in different anatomical positions, which makes it rather difficult to determine and choose the appropriate treatment options. The identification of biomarkers may present clinicians with the ability to determine which treatments will be successful and which may fail due to the development of resistance. One such biomarker informing clinicians what ICI not to use to treat patients is lymphocyte activation gene 3 (LAG-3), a molecule expressed on the surface of and affecting the functionality of regulatory T cells (Tregs).
Melanoma is responsible for nearly 92% of skin cancer-related diagnoses, as well as 60% of skin cancer-related deaths in the United States alone, with no sign of slowing as the diagnosis of melanoma is increasing at a rate greater than that of other cancers [1]. Accumulating mutations in melanin-producing cells or melanocytes causes the rapid expansion and proliferation of a malignant tumor that either remains localized or disseminates and metastasizes throughout the body. Metastatic cancers, such as melanoma, remain incurable for nearly all patients; this is primarily due to the ability of cancer cells to accumulate various mutations, remain unresponsive to chemotherapy, and evade host immune responses [2]. With melanoma cases rising exponentially in recent years, it is crucial to discuss the role of ultraviolet (UV) light (a mutagen), which is a leading stimulus in melanoma development. Long-term exposure to UV light causes melanocytes to accumulate multiple somatic mutations [3]. Due to the ability of mutated melanocytes to proliferate at an uncontrollable rate and metastasize to various anatomical locations within the body, while remaining unresponsive to chemotherapy, finding a cure for melanoma remains a challenging task. Consequently, melanoma accounts for the highest number of skin cancer-related deaths.
Traditional treatments used to fight melanoma include surgery, interferon-alpha 2b, (IFN-α2b), high-dose interleukin-2 (IL-2), and tumor-infiltrating lymphocytes (TIL). Surgery is typically the immediate option for patients diagnosed with melanoma that has not reached the metastatic stage. Being able to excise melanoma without the need for other treatments has made surgery the primary treatment option for this cancer, but it is ruled as an ineffective treatment when patients have reached the advanced stages of the disease. Since surgery is unable to treat melanoma patients at advanced stages, specifically metastatic melanoma (mM), new treatments are being developed and assessed in clinical trials.
The cytokine interferon (IFN) alpha 2b (IFN-α2b) possesses antitumor activities and was used as an immunotherapy regimen for the treatment of melanoma. IFN-α2b received FDA approval for the treatment of melanoma patients who have low-volume disease in their soft tissues [4]. The antitumor effects of IFN-α2b are mediated via caspase-independent apoptosis induction in melanomas [5]. Although IFN-α2b causes a wide range of undesired side effects in stage IV melanoma patients including fatigue, fever, and psycho-cognitive impairment [6], it was considered the first agent to confer significant survival benefits (average survival rate: 3.8 years) in clinical investigations [7].
Interleukin-2 (IL-2) is a T cell growth factor that also inhibits tumor growth [8]. IL-2 was one of the earliest molecules to activate T cells in order to invoke a robust immune response. Since being introduced as a treatment option, IL-2 has led to remission in patients with mM [9]. IL-2 was one of two treatments for mM approved by the FDA; however, clinical trials proved that IL-2 could only induce response rates in 10–20% of patients, ultimately leading to IL-2 being disregarded as a potential cure [10]. Although IL-2 was deemed ineffective when used independently, it has a significant role in new immunotherapeutic options in conjunction with several treatments. One of the most important roles IL-2 played in treating mM was shifting attention towards T cells and their ability to be used in immunity-boosting therapies.
Similar to IL-2, tumor-infiltrating lymphocytes (TIL) shifted attention towards immune system-boosting treatments through the utilization of T cells. The basis for TIL involves isolating T cells that have infiltrated tumor sites and the reinfusion of these naturally occurring T cells into the host to combat cancer cells. TIL therapy consists of three steps—resection, expansion, and reinsertion—in an attempt to invoke robust antitumor immune responses in patients. The first step requires surgical removal of the melanoma and cutting it into small fragments, around 1 mm in size [11]. The identification of activated T cells follows. The process of isolating activated T cells limits the success of TIL due to the difficulty of precisely finding activated T cells in patients diagnosed with mM. Patients diagnosed with the late stages of melanoma have relatively low numbers of activated T cells due to the inherent immunosuppressive properties of the tumor microenvironment. Nevertheless, once this step is completed, TILs will be expanded. T cell expansion occurs over a 28-day period; this timeframe assures adequate activation of T cells; newly expanded T cells will be cryopreserved, followed by reinfusion into the patient [11]. The T cell growth factor, IL-2, is typically used to expand TIL cultures. Activated and expanded TIL cultures will be reinfused back into patients. However, prior to reinfusion, patients receive a lymphodepleting regimen in order to prepare the microenvironment for reinfused TILs. Lymphodepletion increases objective tumor regression in nearly 50–70% of patients, even leading to complete regression in some patients [12]. Although TIL therapy has demonstrated promising results and can be used in treating other cancers such as renal carcinoma, it has been ruled as ineffective as TILs are typically being generated from half of all collected samples [13]. In addition, TIL requires patients to have T cells that are already activated and have penetrated the tumor microenvironment. These difficulties have led to an undeniable need for new therapies to treat mM.
The MAPK (Mitogen Activated Protein Kinase) signal transduction pathway controls the proliferation, differentiation, and survival of the cells. The ERK1/2 (extracellular signal-regulated kinase) signaling pathway is the major MAPK signaling pathway, mainly involved in the proliferation and survival of tumor cells, leading to their growth [14]. The components of the ERK1/2 pathway include the GTPase RAS, which, upon activation, activates the RAF molecules (e.g., ARAF, BRAF, CRAF) and subsequently phosphorylates MEK and ERK kinases. BRAF is principally activated by RAS and, once mutated, acts as a monomer-independent stimulus ( Figure 1 A) [15][16][17][18][19].
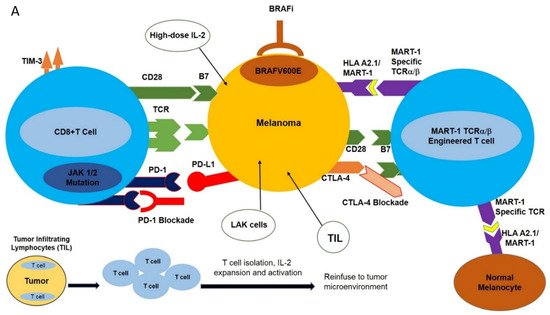
Approximately 60% of melanoma patients harbor various BRAF mutations; 90% of these mutations are substitutions of glutamic acid for valine at position 600; BRAF V600E , leading to constitutive MAPK activation, tumor proliferation, and a significant (500X) increase in BRAF kinase activity compared to wild-type BRAF [20][21]. The oral serine-threonine kinase inhibitor called vemurafenib (PLX4032, RG7204) is a BRAF V600E -specific inhibitor that inhibits the MAPK pathway and MEK and ERK phosphorylation, induces cell cycle arrest, and activates apoptotic pathways in BRAF V600E -mutated cells [19].
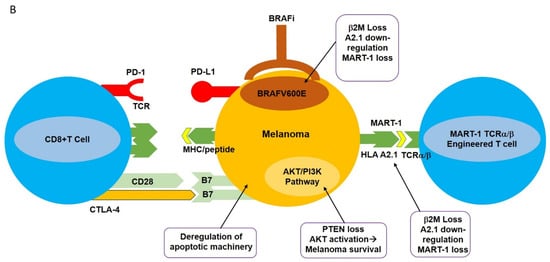
Vemurafenib has improved the survival rate in patients with metastatic melanoma harboring BRAF V600E mutations. After successful in vitro and in vivo experiments, vemurafenib is considered a promising drug for use against melanoma (discussed below). However, melanomas treated with vemurafenib will eventually adopt various resistance mechanisms that progress over time, leading to eventual relapse [20][21][22].
The MEK inhibitor AZD6244 efficiently inhibits the MAPK pathway; however, its benefits are often offset as it impairs T cell function [23]. Another MEK inhibitor called PD0325901 produced a significant decrease in ERK phosphorylation and led to disease stabilization in phase I clinical trials ( Figure 1 A) [24][25].
Monoclonal antibody-mediated blockade of immune checkpoints such as PD-1/PD-L1 and CTLA-4, as well as targeted inhibition of BRAF V600E , have had profound results in various clinical trials. However, the clinical utilization of these modalities is hampered by the development of resistance as well as limited response rates. This has spurred the design of new strategies with high selectivity towards melanomas including adoptive cell transfer (ACT). This new immunotherapy-based treatment involves ex vivo expansion and activation of HLA A2.1/MART-1-specific T cell receptor (TCR) engineered T cells and reinfusion of these newly expanded high-affinity MART-1-specific T cells into patients with metastatic melanoma [26]. Heterodimeric T cell receptors (TCR) consist of two transmembrane polypeptide chains, one alpha chain, and one beta chain. Each chain devotes a portion of itself for stable attachment on the T cell surface, and a variable region responsible for binding to the Major Histocompatibility Complex (MHC) [1]. TCRα/β-engineering gives T cells high specificity and increased affinity for the MART-1 melanoma-specific antigen expressed in the context of HLA A2.1 ( Figure 1 A) [27].
While traditional ACT has shown promise, modifications have been made to the protocol to increase the efficacy of treatment. These fine-tunings include the pre-infusion of a lymphodepleting regimen, and postinfusion use of high-dose IL-2. Lymphodepletion prior to the infusion of T cells has been proven to promote the antitumor response. This is possibly because of its ability to promote an environment that is cleared of potentially inhibiting molecules [28]. Lymphodepletion has been proven to promote the recruitment of homeostatic cytokines IL-15 and IL-17 to the tumor milieu in clinical trials [29], and also provides the physical space for TCR transgenic T cells. IL-2 has the innate ability to promote T cell growth; therefore, its postinfusion inclusion in clinical settings of TCR-engineered T cell therapy of melanoma ensures the sustained expansion and activation of infused MART-1 TCR T cells.
Overall, TCR-engineered melanoma immunotherapy has led to varying responses, ranging from being ineffective to complete remission. TCR-engineered ACT will thus continue to play a significant role in cancer immunotherapy. Due to its specificity and high affinity for tumor-associated antigens, combined with its potential efficacy, TCR-engineered T cell ACT remains a preferred treatment option [30]. However, TCR-engineered T cell therapy protocols require further optimization to increase the sustainability and efficacy of genetically modified T cells in the treatment of metastatic cancers including melanoma. Combination of TCR-engineered T cell ACT with ICIs to increase treatment efficacy may be a plausible area of future investigation [31].
Aside from CRS, autoimmune responses pose a tremendous challenge for patients receiving ACT treatment with TCR-engineered T cells. Autoimmune responses are the result of “On Target, Off Tumor” phenomena. This “ On Target, Off Tumor” response results from newly infused T cells attacking the correct antigens, but in the wrong locations. Tumor-associated antigens (TAA) are expressed not only on tumors but also on naturally occurring, nonmutated cells [2]. There is currently no strategy that can ensure newly infused TCR-engineered T cells will only target mutated/overexpressed tumor-associated antigens, as was demonstrated for MART-1-specific T cells. The newly infused TCR-engineered T cells were expected to attack the MART-1/HLA A2.1 complex on melanomas. However, it was later discovered that MART-1 antigens are not only expressed on melanomas, but also on naturally occurring melanocytes [2]. Patients who received this treatment typically experienced vitiligo in one or more locations. Similarly, in the context of clinical utilization of MAGE-A3-specific T cells, at the time of treatment it was unclear whether MAGE-A3 was also expressed in the human brain. Postclinical trial results included five patients showing tumor regression, three patients experiencing neurotoxic effects, and two patients who had died [32]. These clinical results were due to the newly infused MAGE-A3 TCR transgenic T cells attacking the MAGE-A3 antigens expressed in the brain.





