This paper describes the development of a simple voltammetric method for the measurement of 2,4-dinitrotoluene (24DNT) at glassy carbon electrode (GCE). Initial investigations were undertaken using cyclic voltammetry to characterise the redox behaviour of 24DNT. Over the pH range 2 to 10 two pH dependent reduction peaks were recorded on the initial negative going scan, concluded to result from the reduction of the two nitro groups to the corresponding hydroxylamines. On the return positive going scan two oxidation peaks were recorded, resulting from the oxidation of the hydroxylamine (O1) formed on the initial negative scan. At pH 6, the peak potential of the oxidation process O1 occurred at a potential close to 0 V and was chosen for investigation. The optimum voltammetric conditions required were identified to be supporting electrolyte of 0.1 M pH 6.0 phosphate buffer containing 10 % acetonitrile. Using differential pulse voltammetry, the calibration plot was found to be linear from 5.0 µM to 500 µM (R2= 0.9979), with a detection limit of 11.0 µM (based on a signal-to-noise ratio of 3).
- Cyclic voltammetry
- Differentail pulse voltammetry
- 2,4-dinitrotoluene
- glassy carbon electrode
- mechanism
Introduction
The detection of explosives and their residues is important both in conventional and asymmetric warfare and for the detection of terrorist and insurgent activity. Nitro compounds such as trinitrotoluene (TNT), RDX or tetryl are commonly employed both by the military in mines, shells and other ordnance and by terrorists in improvised explosive devices. These can be difficult to detect using approaches such as metal detectors, as they can have very little metal in their construction or their signature is obscured by other un-associated metal objects. Present attempts for their detection have therefore focused on alternative methods such as the determination of the explosive utilised or its vapour.
Commonly used nitro based explosives have very low vapour pressures and hence are not generally good candidates for detection via this approach. However, these can contain a number of more volatile compounds such as 2,4-dinitroluene (24DNT) which can be employed as a possible marker for their detection. Consequently, the development of devices capable of determining low concentrations of 24DNT in vapour is highly important. In addition to this, 24DNT is also utilised for the manufacture of furniture foam and dyes and environmental and industrial monitoring of pollution and exposure is also an important consideration as 24DNT exposure has been associated with lung, liver and reproductive problems.[1]
Aromatic nitro-compounds, such as 24DNT have been shown to exhibit a rich electrochemical which has been noted by ourselves[2][3][4][5][6][7] and other researchers.[8][9][10][11][12][13][14][15] Therefore, we believe it possible to use a similar approach for the determination of 24DNT both initially in solution and in the vapour phase. The majority of previously reported electrochemical methods for the detection of nitroaromatic compounds have utilised the direct electrochemical reduction of the nitro group to either its corresponding hydroxylamine (eq. 1) or amine as the analytical signal. However, in this present proof of concept study, we have investigated the alternative possibility of determining 24DNT via the subsequent oxidation of the electrochemically generated hydroxylamine to its corresponding nitroso species (eq. 2).
Ar-NO2 + 4e‑ + 4H+ → Ar-N(H)OH + H2O eq.1
Ar-N(H)OH → Ar-N=O + 2e‑ + 2H+ eq.2
At more negative applied potentials it is possible to reduce the hydroxylamine further to the corresponding primary amine, as described in equation 3.
Ar-N(H)OH + 2e‑ + 2H+ → Ar-NH2 + H2O eq.3
We believe that the oxidation process described in eq.2 offers a number of advantages, as it this process as it is removed from possible common interferences, such as the reduction of oxygen [6] and occurs at low applied potentials, with corresponding low background currents, leading to improved detection limits.
In the first section of this study, cyclic voltammetric studies were made to optimise the electrochemical conditions required for the determination of 24DNT in the liquid phase. We next explored and optimised the conditions required for the determination of 24DNT employing the more sensitive differential pulse voltammetric (DPV) waveform.
Experimental
Chemicals and Reagents
All chemicals were obtained from Fischer (Loughborough, UK), unless otherwise stated. Deionised water was obtained from a Purite RO200-Stillplus HP System, (Purite Oxon., UK). Stock solutions of sodium orthophosphate were made at a concentration of 0.2 M by dissolving the appropriate mass of solid in deionised water to give a 0.2 M solution. A stock solution of orthophosphoric acid was made by dilution in deionised water. These were then titrated together to give the desired pH. Stock solutions of 2,4-dinitrotoluene were prepared by dissolving the required mass in acetonitrile to give a concentration of 10 mM. Working standards, for voltammetric studies, were prepared by dilution of this primary stock solution with phosphate buffer and acetonitrile to give a final concentration of 0.1 M phosphate buffer and 10 % acetonitrile via the addition of sufficient deionised water.
Apparatus
Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were undertaken using an Ivium CompactStat potentiostat (Ivium Technologies, The Netherlands) interfaced to a PC for instrument control and data acquisition. The voltammetric cell contained a platinum wire auxiliary electrode, a Ag/AgCl reference electrode, and a glassy carbon electrode (GCE) (3 mm diameter), as the working electrode. The GCE was cleaned before each scan by polishing with slurry of aluminium powder. Cyclic voltammograms were initially recorded with plain solutions of 0.1 M phosphate buffer, containing 10 % acetonitrile and then in the same solution containing 1 mM 24DNT. A starting and end potential of +1.0 V and a switching potential -1.5 V were utilised. Differential pulse voltammetry was undertaken using a starting potential of ‑0.7 V and a final potential of 0.0 V; using a step height of 10 mV, pulse repetition time 0.2 s, pulse amplitude of 75 mV, and pulse duration of 50 ms.
Results and Discussion
Voltammetric Studies of 2,4-Dinitrotoluene at Glassy Carbon Electrode
Initial cyclic voltammetric studies were made with a 1 mM solution of 24DNT, dissolved in 0.1 M phosphate buffer containing 10 % acetonitrile (10 mL). Figure 1 shows the resulting cyclic voltammogram obtained using a scan rate of 50 mV/s. This shows the appearance of two reduction peaks, R1 and R2 on the first forward scan. These two reduction peaks were concluded to result from the formation of hydroxylamine moieties (eq. 1) formed form the sequential reduction of the two nitro groups. On the return scan, an oxidation process, O1 was recorded. The oxidation O1 was considered to be the result of oxidation of the hydroxylamine species formed on the initial negative going scan to give the corresponding nitrosamine (eq. 2). If the scan was repeated, a third reduction peak, R3 was recorded, resulting from the reduction of the nitroso species formed at O1 to a hydroxylamine (the reverse of equation 2).
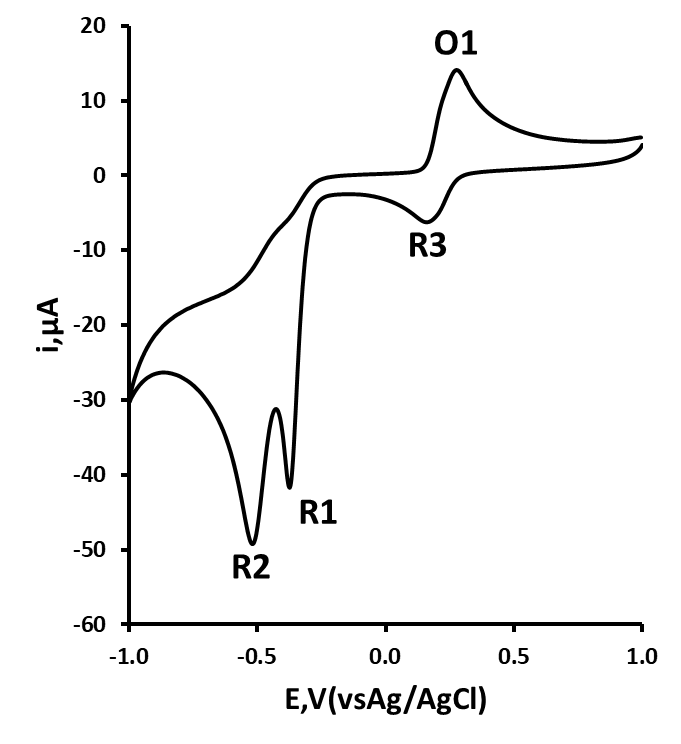
Figure 1. Cyclic voltammogram (second scan), obtained at a scan rate of 50 mV/s, for a 1 mM solution of 2,4-dinitrotoluene in 10 % acetonitrile, buffered with 0.1 M pH 2.0 phosphate. Starting and end potential; +1.0 V, switching potential ‑1.0 V.
Further investigations were undertaken into characterising the oxidation process recorded as O1. By differential pulse voltammetry (DPV). This waveform is known to exhibit both greater sensitivity and selectivity, being able to discriminate between faradic and capacitance currents and processes with similar reduction and oxidation potentials (Eps) than the linear sweep type waveform used in cyclic voltammetry. Using DPV, we were able to discern that the process O1 was in fact two oxidation peaks with very similar Eps values. We believe that is occurs as a result of the oxidation of the two possible hydroxylamine compounds formed at R1 and R2.
Effect of pH
For most organic compounds both electrons and protons are involved in their redox reactions. As a results the peak potential (Ep) can be seen to be related to the pH of the supporting electrolyte. The relationship can be described by the following equation (eq.4): [16]
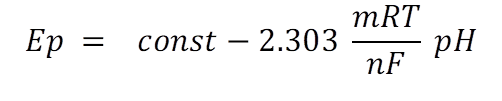 eq.4
eq.4
where R is the gas constant (8.314 J K‑1 mol‑1), T is the temperature (K), F is the Faraday constant (96 485 C mol‑1), and m and n are the number of protons and electrons involved in the oxidation, respectively. From eq.4, it can be seen that Ep varies by 59(m/n) mV per pH unit.
Figure 2 shows the plots of Ep versus pH for peaks R1, R2, R3 and O1 obtained over the range pH 2–11. The oxidation peak O1 (Figure 2a) was found to be pH dependant over the entire pH range studied.[17] A plot of Ep vs. pH was found to give a slope of 59 mV/pH, indicative of an equal number of protons and electrons involved in the oxidation step. The reduction peaks R1 and R2 are pH dependent over the pH range 2 to 8 with slopes of 90 mV/pH between pH 2 and 4 and 45 mV/pH from pH 4 to pH 8 corresponding to a reduction process involving 3 electrons and 4 protons.
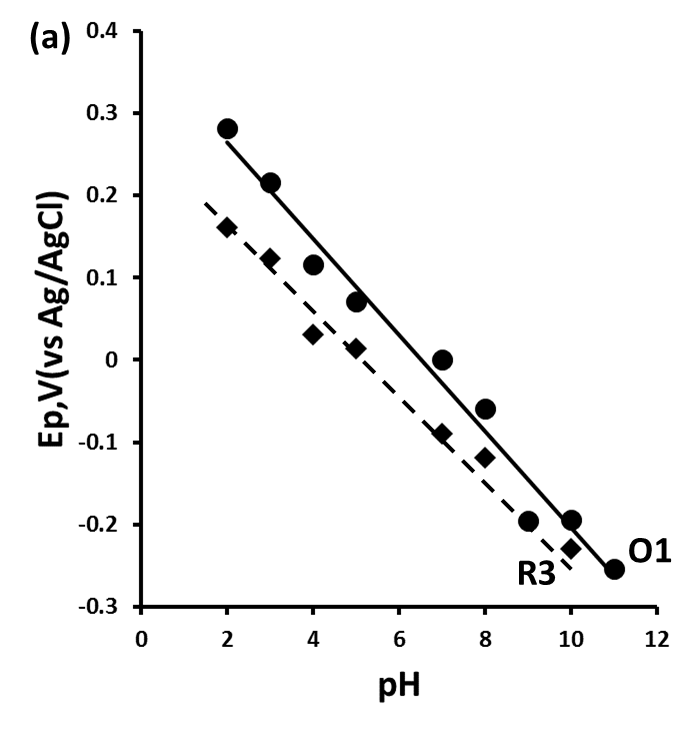
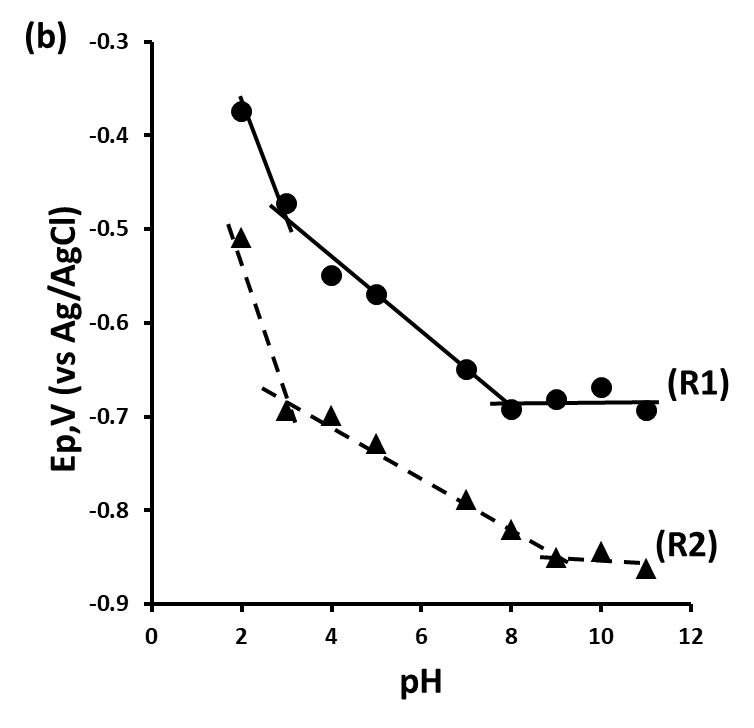
Figure 2. Plot of Ep and ip vs. pH for 2,4-dinitrotoluene. Ep vs. pH (a) O1 and R3, (b) R1 and R2.
At pH values below pH 4, the nitro group is in its fully protonated from, Ar-NO2H2 .[18] This allows for it to be reduced in a 4e‑, 2H+ reduction to the hydroxylamine rather than four protons required at higher pH values (eq.5). Similarly, at pH values between pH4 and pH8, the mono-protonated form, Ar-NO2H, allows for a 4e‑, 3H+ reduction step (eq.6), shown in the corresponding change in slope seen in figure 2.
Ar-NO2H2 + 4e‑ + 2H+ → Ar-N(H)OH + H2O eq.5
Ar-NO2H + 4e‑ + 3H+ → Ar-N(H)OH + H2O eq.6
At pH values lower than 3, the slope of the Ep vs. pH was found to increase to 90 mV/pH unit, indicative of a 3H+, 2e‑ reduction process. We believe that this is a result of the further reduction of the nitro group to the corresponding primary amine (eq.7).
Ar-N(H)OH + 2e‑ + 2H+ → Ar-NH2 + H2O eq.7
Investigations have shown the dissociation constant of the protonated radical anion for nitrobenzene to be pK; = 3.2 and for substituted nitrobenzenes values between pK, = 2.2 and pK, = 3.9 have been reported[19] close to the pH value in which the slopes of the Ep vs pH change in figure 2.
At pH values greater than pH 8, the Ep values for both R1 and R2 became independent of pH. This is consistent with that reported by Sadek and Abd-El-Nabey who showed similar behaviour for the polarographic reduction of nitrobenzene. With supporting electrolytes with pH values above 8 the low concentrations of protons allows for the reduction of the nitro group to occur via a two electron reduction step as described in equation 8.
Ar-NO2 + 2e‑ → Ar-NO22‑ eq.8
This can then undergo a 2 electron oxidation back to corresponding nitro group.
On the return positive going scan an oxidation process (O1) is recorded, resulting from the oxidation of the hydroxylamine formed on the previous negative going scan. This oxidation signal is believed to result from the oxidation of the hydroxylamine to the corresponding nitroso species (eq.2). However, it is evident that this is not a single peak and is actual comprised of two peaks with very similar Ep values. This is similar to that previously described for the cyclic voltammetric behaviour of 3,5-dinitrobenzoic acid.[20]
The peak current (ip) of all three peaks were found to be pH dependent over the range studied (Figure 2). We were practical interested in the oxidation process seen as the peak O1. As this occurred at a potential close to 0 V it was chosen to investigate this peak further as this offers a number of analytical advantages; such as lower background currents and is free from common interference such as oxygen.
Effect of Scan Rate
The effect of scan rate was studied on the cyclic voltammetric behaviour of 24DNT was investigated over the range of 20 to 200 mV/s for each pH point investigated. A plot of peak height vs. the square root of scan (v½) was found to linear for all the peaks studied indicating a diffusional process.
Effect of Starting Potential
For further studies it was decided to use the differential pulse waveform as it offers advantages over the linear sweep waveform; such as improved discrimination between faradaic current and capacitance current giving leading to better sensitivity and resolution. Studies were now made into investigating the effect of starting potential on the result ip magnitude of O1 using a pre-treatment time of 20 s at pH 6. As can be seen in figure 3, a sudden rise in the resulting peak current is seen at applied potentials more negative than ‑0.4 V. At potentials more negative than ‑0.6 V (vs. Ag/AgCl) the resulting peak current plateaus and becomes constant with increasing negative potentials. Consequently, further studies were made using an applied potential of ‑0.7 V (vs. Ag/AgCl).
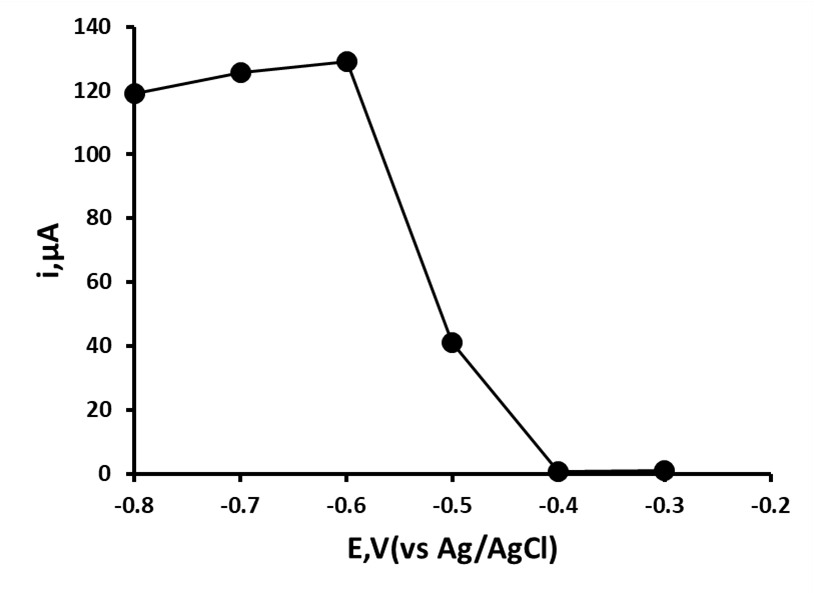
Figure 3. Effect of starting potential on 0.1 mM 2,4-dinitrotoluene O1 oxidation peak obtained by differential pulse voltammetry at pH 6.0.
Calibration Curve and Limit of Detection
Initial studies were undertaken to study the effect of 24DNT concentrations on the magnitude of the differential pulse peak. The calibration plot was found to be linear from 5.0 µM to 500 µM (R2 = 0.9979), with a detection limit of 11.0 µM (based on a signal-to-noise ratio of 3) and quantification limit of 112 µM (based on a signal-to-noise ratio of 10).
Conclusion
This is believed to be the first report on the utilisation of the redox generated hydroxylamine for the determination of 24DNT. The paper demonstrates that 24DNT yields a well-defined voltammetric signal at a GCE. A simple and convenient assay for 24DNT was developed requiring only the dilution of sample in the optimised supporting electrolyte. A detection limit of 98.4 ng/mL with a linear response from 11 µM up to 500 µM (R2 = 0.9979). This approach avoids the problems associated with the high positive or negative potentials normally used for the determination of 24DNT.
References
- Honeychurch, K. C. and Hart, J. P.; Voltammetric behaviour of p-nitrophenol and its trace determination in human urine by liquid chromatography with a dual reductive mode electrochemical detection system. Electroanalysis 2007, 19, 2176-2184, doi.org/10.1002/elan.200703989|.
- K.C Honeychurch; J.P Hart; P.R.J Pritchard; S.J Hawkins; N.M Ratcliffe; Kevin Honeychurch; Development of an electrochemical assay for 2,6-dinitrotoluene, based on a screen-printed carbon electrode, and its potential application in bioanalysis, occupational and public health. Biosensors and Bioelectronics 2003, 19, 305-312, 10.1016/s0956-5663(03)00208-2.
- Honeychurch K.C.; Development of an electrochemical assay for the illegal “fat burner” 2,4-dinitrophenol and its potential application in forensic and biomedical analysis. Advances in Analytical Chemistry 2016, 6, 41-48, 10.5923/j.aac.20160602.02 .
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for 2,4-Dinitrotoluene and 2,6-Dinitrotoluene (Update); Public Health Service, U.S. Department of Health and Human Services, : Atlanta, GA. USA, 1998; pp. p.
- Kevin C. Honeychurch; Joshua Brooks; John P. Hart; Development of a voltammetric assay, using screen-printed electrodes, for clonazepam and its application to beverage and serum samples. Talanta 2016, 147, 510-515, 10.1016/j.talanta.2015.10.032.
- Kevin C. Honeychurch; Gwen M. Davidson; Emma Brown; John P. Hart; Novel reductive–reductive mode electrochemical detection of Rohypnol following liquid chromatography and its determination in coffee. Analytica Chimica Acta 2015, 853, 222-227, 10.1016/j.aca.2014.09.033.
- Kevin C. Honeychurch; Adrian Crew; Hannah Northall; Stuart Radbourne; Owian Davies; Sam Newman; John P. Hart; The redox behaviour of diazepam (Valium®) using a disposable screen-printed sensor and its determination in drinks using a novel adsorptive stripping voltammetric assay. Talanta 2013, 116, 300-307, 10.1016/j.talanta.2013.05.017.
- Min-Chieh Chuang; Joshua Ray Windmiller; Padmanabhan Santhosh; Gabriela Valdés Ramírez; Michal Galík; Tzu-Yang Chou; Joseph Wang; Santhsoh Padmanabhan; Textile-based Electrochemical Sensing: Effect of Fabric Substrate and Detection of Nitroaromatic Explosives. Electroanalysis 2010, 22, 2511-2518, 10.1002/elan.201000434.
- Juliane R. Sempionatto; Rupesh K. Mishra; Aida Martín; Guangda Tang; Tatsuo Nakagawa; Xiaolong Lu; Alan S. Campbell; Kay Mengjia Lyu; Joseph Wang; Wearable Ring-Based Sensing Platform for Detecting Chemical Threats. ACS Sensors 2017, 2, 1531-1538, 10.1021/acssensors.7b00603.
- Kouadio Fodjo Essy; Li Yuan-Ting; Li Da-Wei; Sara Riaz; Long Yi-Tao; Rapid determination of nitrophenol isomers in polluted water based on multi-walled carbon nanotubes modified screen-printed electrode. Mediterranean Journal of Chemistry 2011, 1, 19-29, 10.13171/mjc.1.1.2011.08.06.23.
- Krausa, M., Doll, J., Schorb, K., Böke, K., Hambitzer G.,; Fast electrochemical Detection of Nitro‐ and Aminoaromates in Soils and Liquids. Propellants, Explosives, Pyrotechnics 1997, 22, 156-159, doi.org/10.1002/prep.19970220311.
- Karel Cizek; Chad Prior; Chongdee Thammakhet; Michal Galík; Kevin Linker; Ray Tsui; Avi Cagan; John Wake; Jeff La Belle; Joseph Wang; et al. Integrated explosive preconcentrator and electrochemical detection system for 2,4,6-trinitrotoluene (TNT) vapor. Analytica Chimica Acta 2010, 661, 117-121, 10.1016/j.aca.2009.12.008.
- Ti-Wei Chen; Zhen-Huan Sheng; Kang Wang; Feng-Bin Wang; Xing-Hua Xia; Determination of Explosives Using Electrochemically Reduced Graphene. Chemistry - An Asian Journal 2011, 6, 1210-1216, 10.1002/asia.201000836.
- Lourdes Agüí; Denis Vega-Montenegro; Paloma Yáñez-Sedeño; José M. Pingarrón; Rapid voltammetric determination of nitroaromatic explosives at electrochemically activated carbon-fibre electrodes. Analytical and Bioanalytical Chemistry 2005, 382, 381-387, 10.1007/s00216-004-3017-z.
- Joseph Wang; Ron K Bhada; Jianmin Lu; Douglas Macdonald; Remote electrochemical sensor for monitoring TNT in natural waters. Analytica Chimica Acta 1998, 361, 85-91, 10.1016/s0003-2670(97)00702-2.
- Qianqi Lin; Qian Li; Christopher Batchelor-McAuley; Richard G. Compton; Two-Electron, Two-Proton Oxidation of Catechol: Kinetics and Apparent Catalysis. The Journal of Physical Chemistry C 2015, 119, 1489-1495, 10.1021/jp511414b.
- H. Sadek; B.A. Abd-El-Nabey; Polarography of nitrobenzene in aqueous solutions and in water-alcohol mixtures. Electrochimica Acta 1972, 17, 2065-2075, 10.1016/0013-4686(72)80030-6.
- D. Dumanovic; J. Jovanovic; D. Sužnjević; M. Erceg; P. Zuman; Polarographic and electrochemical studies of some aromatic and heterocyclic nitro compounds, part III: Electroreduction of mono- and dinitropyrazoles and -imidazoles. Electroanalysis 1992, 4, 871-887, 10.1002/elan.1140040908.
- Petr Zuman; Zbigniew Fijalek; Dragica Dumanović; Desanka Sužnjević; Polarographic and electrochemical studies of some aromatic and heterocyclic nitro compounds, part I: General mechanistic aspects. Electroanalysis 1992, 4, 783-794, 10.1002/elan.1140040808.
- R. Moscoso; J. Carbajo; J.D. Mozo; J.A. Squella; Voltammetric behavior of 3,5-dinitrobenzoic acid in solution on GCE and encapsulated on multiwalled carbon nanotube modified electrode. Journal of Electroanalytical Chemistry 2016, 765, 149-154, 10.1016/j.jelechem.2015.08.010.

