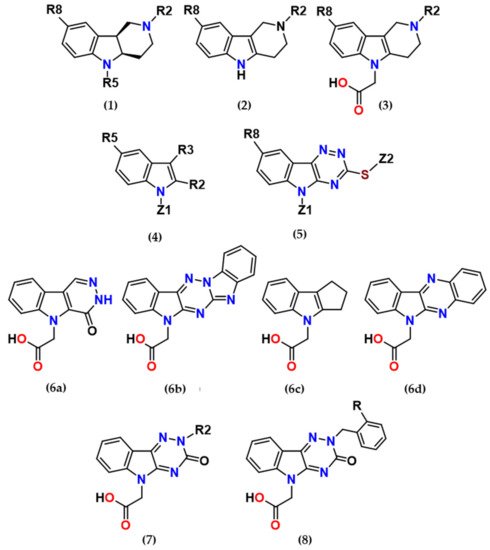
With regard to the multifactorial pathophysiological origin of diabetic complications, a therapeutic approach based on the use of multi-target directed drugs has been forwarded. Based on the premise that a bifunctional compound with joint antioxidant/aldose reductase inhibitory (AO/ARI) activities could be multifactorially beneficial, we were inspired by both an efficient antioxidant stobadine (1a), a drug of hexahydropyridoindole nature, and by the highly efficient ARI lidorestat, derivative of indol-1-yl acetic acid. Stobadine (1a) as an efficient ROS scavenger was extensively studied in multiple models of diabetic complication with the aim to attenuate the oxidative component of glucose toxicity. Lidorestat belongs to a broad class of acidic aldose reductase inhibitors with characteristic carboxymethyl pharmacophore.
At the very beginning of the drug design, we employed the hexahydropyridoindole moiety of stobadine (1a) as a starting scaffold, which was sequentially endowed with the carboxymethyl pharmacophore in three different synthetically accessible positions (2, 5 and 8). SAR study of the novel derivatives in relation to the carboxymethyl pharmacophore regioisomerization and core scaffold modification resulted in developing of
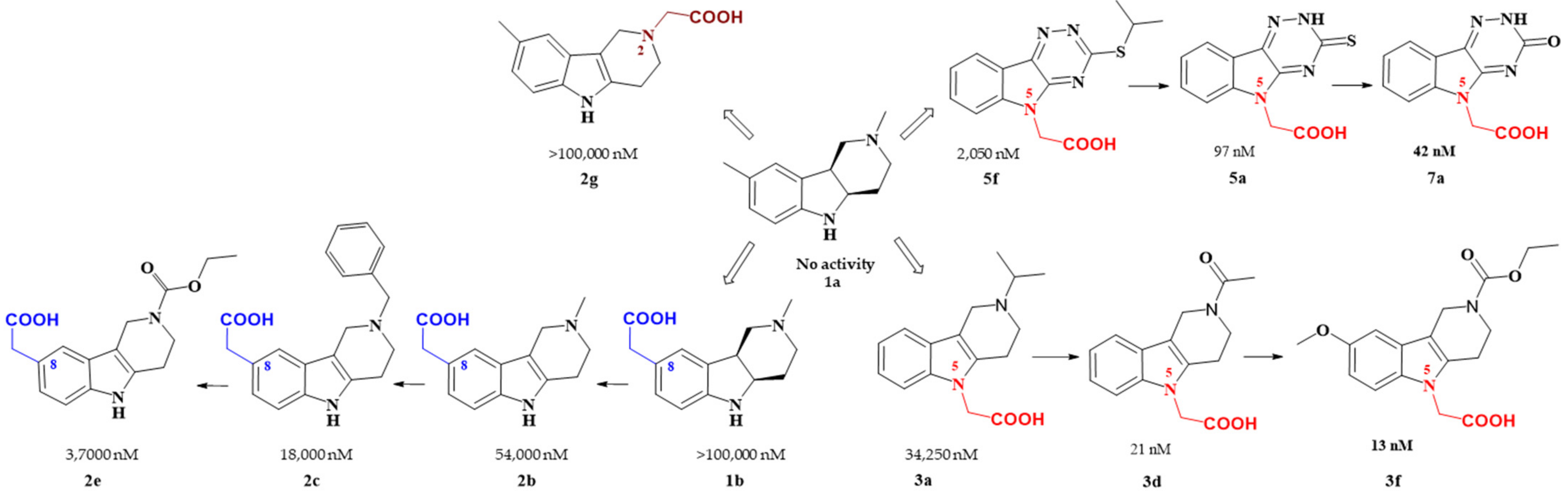
several promising series of aldose reductase inhibitors/antioxidants as summarized in Figure 2.
Figure 2. Carboxymethyl pharmacophore regioisomerization in AR inhibitory activity optimization starting from the non-active hexahydropyridoindole scaffold 1a
Starting from the efficient hexahydropyridoindole antioxidant stobadine (1a), two series of hexahydro- and tetrahydropyridoindoles carboxymethylated in position 8 were synthesized and characterized as AR inhibitors with negligible (e.g. compound 1b) or mild (e.g. compounds 2b,c) efficacy and selectivity yet with significant antioxidant (AO) effect as an additional biological activity. The hexahydropyridoindole scaffold was excluded from further drug design since the marked antioxidant activity of the hexahydropyridoindoles was combined with only minor AR inhibition (e.g. compound 1b) which was explained by space distortion of the hexahydropyridoindole structure not fitting properly the AR binding pocket. Tetrahydropyridoindole congeners with a planar tricyclic moiety appeared more promising route in designing efficient ARIs. Notably, elimination of the basicity center at N2 position, which prevented zwitterions formation, significantly improved AR inhibition as shown in the case of compound 2e.
Shifting the carboxymethyl pharmacophore from position 8 to position 2, yielded derivatives with markedly decreased inhibition efficacy, as exemplified by compound 2g, therefore this route of drug designing was not further followed.
Structure optimization of the tetrahydropyridoindole scaffold by transferring the carboxymethyl pharmacophore from position 8 to position 5, yielded derivatives with markedly enhanced inhibition efficacy and selectivity, yet with abolished AO activity (e.g. compounds 3a,d,f). In this series, the AR inhibition efficacy markedly increased with decreasing basicity of N2 nitrogen as documented by compound 3f.
Thioxotriazine structural alternatives yielded highly efficient AR inhibitors (e.g. compound 5a) with high selectivity and reasonable AO activity. In further structure optimization efforts, isosteric replacement of sulfur in compound 5a with oxygen provided compound 7a with increased AR inhibition efficacy and markedly improved selectivity, yet with diminished AO activity.
Interestingly, the structural features of the most efficient ARIs resulting from the above optimization procedure, namely compounds 2c, 3f, 5a and 7a (IC50(ALR2) = 18 000; 13; 97; 42 nM, resp.) still match the strict criteria of the „rule of three“, which points to their excellent “leadlikeness” with prospects of further structure optimizations.
Among the novel derivatives, compound
5a was patented (P3). At the present time, it is in the stage of complex preclinical evaluation under the name of cemtirestat [
76,
78,
87,
88,
89,
94,
95,
96,
97,
98,
99,
100]. Apparently, there are several advantages of cemtirestat over the clinically used epalrestat, namely lower molecular weight, better water solubility, higher AR inhibition activity recorded both at the level of isolated enzyme ( and ) and at the organ level of isolated rat eye lenses () and additional AO action ( and , and ). Recently, antioxidant activity of cemtirestat was corroborated by a study of Valachova et al. [
96] reporting ability of the drug to protect hyaluronan against oxidatively induced degradation. In addition, protective effects of cemtirestat in neuron-like PC12 cells and BV2 rodent microglial cells exposed to toxic models of oxidative stress were recently documented [
97,
98]. Studies in vivo in rat models of diabetes revealed ability of cemtirestat (
5a) to attenuate symptoms of peripheral neuropathy with high significance [
89,
95,
100]. Moreover, our very recent study [
89] proved that the direct ROS scavenging activity of cemtirestat (
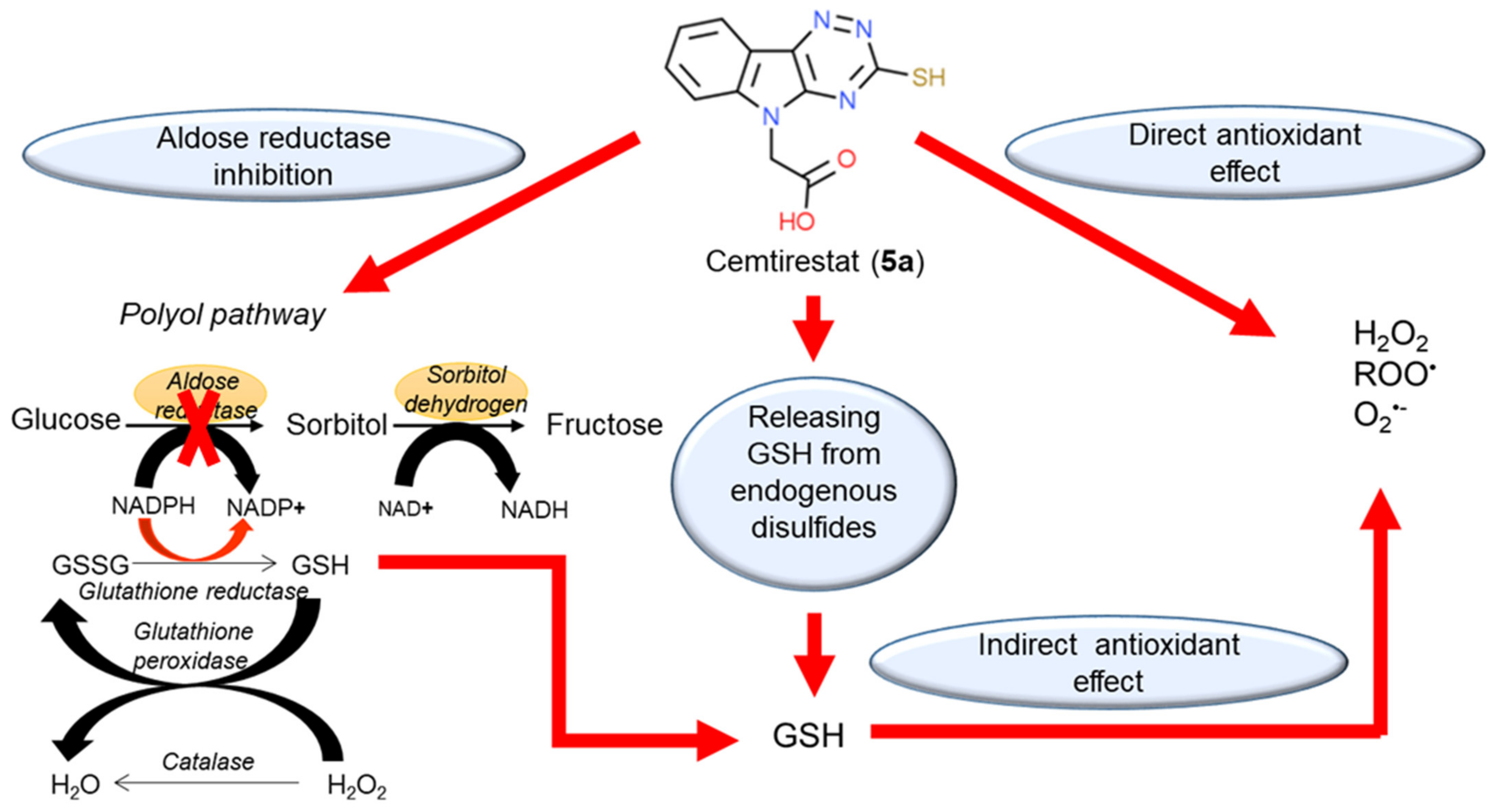
5a) is complemented by its ability to restore thiol-disulfide homeostasis by releasing free GSH from the pool of endogenously bound disulfides. In addition, ceasing aldose reductase activity by cemtirestat spares NADPH, which is needed for recycling of GSH by glutathione reductase. Considering the above-mentioned complementary activities (), cemtirestat (
5a) represents a practical example of a therapeutic strategy against chronic complications in diabetes based on multiple pharmacological activities. Yet, in-depth studies of the indicated molecular mechanisms and their mutual interactions are still needed.
Figure 3. Multiple pharmacological activities of cemtirestat (
5a). Reprinted with permission from [
89]. Copyright (2020) Elsevier.
3. Patents
1. Štefek, M.; Šnirc, V.; Demopoulos, V.; Djoubissie, P.; Račková, L.; Májeková, M.; Karasu, C. Carboxymethylated pyridoindoles as pharmaceutical agents, 8.6.2009, Slovak Patent No. 28695.
2. Štolc, S.; Považanec, F.; Bauer, V.; Májeková, M.; Wilcox, A.; Šnirc, V.; Račková, L.; Sotníková, R.; Štefek, M.; Gáspárová-Kvaltínová, Z.; Gajdošíková, A.; Mihálová, D.; Alföldi, J. Pyridoindole derivatives with antioxidant properties, preparation and therapeutic use, 7.12.2010, Slovak Patent No. 287506.
3. Štefek, M.; Miláčková; Diez-Dacal, B.; Pérez-Sala, D.; Šoltésová Prnová, M. Use of 5-carboxymethyl-3-mercapto-1,2,4-triazino-[5,6-b]indoles and their pharmaceutical composition, 3.4.2015, WO2015057175; 3.10.2017 Slovak Patent No. 288508.
4. Štefek, M.; Ballekova, J.; Šoltesová-Prnová, M.; Májeková, M. Use of 5-Carboxymetyl-1,2,3,4-tetrahydro-1H-pyrido[4,3-b] indoles and their pharmaceutical composition, 7.1.2020, Slovak Patent No. 288725.
5. Štefek, M.; Kováčiková, L.; Šoltésová Prnová, M.; Addová, G.; Boháč, A. Cemtirestat disulfide prodrug of aldose reductase inhibitor, preparation, pharmaceutical composition and therapeutic use, 14.12.2020, Slovak Patent Application No. 50074-2020.
(References would be added automatically after the entry is online)