

Desmin intermediate filaments (IFs) is one of cytoskeleton components of muscle cells and play an important role in maintaining their structural and functional integrity .Disturbance of their network due to desmin mutations or deficiency leads to an infringement of myofibril organization and to a deterioration of mitochondrial distribution, morphology, and functions. The nature of the interaction of desmin IFs with mitochondria is not clear. To elucidate the possibility that desmin can directly bind to mitochondria, we have undertaken the study of their interaction in vitro. Using desmin mutant Des(Y122L) that forms unit-length filaments
(ULFs) but is incapable of forming long filaments and, therefore, could be effectively separated from mitochondria by centrifugation through sucrose gradient, we probed the interaction of recombinant human desmin with mitochondria isolated from rat liver. Our data show that desmin can directly bind to mitochondria, and this binding depends on its N-terminal domain. We have found that mitochondrial cysteine protease can disrupt this interaction by cleavage of desmin at its N-terminus.
- mitochondria
- desmin
- intermediate filaments
- calpain
Note:All the information in this draft can be edited by authors. And the entry will be online only after authors edit and submit it.
1. Introduction
Intermediate filament (IF) networks are one of three cytoskeletal components along with microtubules and actin microfilaments. IFs provide mechanical strength and elasticity to cells and tissues and regulate intracellular positioning of nucleus and organelles [1]. It has been shown that one of the important roles of IFs inside animal cells is to ensure normal mitochondrial functioning [2–7]. Desmin, a major constituent of the IF network inside muscle cells, is a good example of such factors controlling mitochondria. A plethora of pathological conditions, collectively known as desminopathies, is caused by mutations in desmin and results in abnormalities in mitochondrial distribution and morphology as well as reduced mitochondrial respiratory function [5,7–9]. These data suggest that interaction with desmin is a prerequisite for normal mitochondrial functioning. However, the nature of interaction between desmin IFs and mitochondria is not fully elucidated. For example, one question that needs to be asked is whether desmin is able to bind directly to these organelles or some intermediary proteins are necessary. Previously, in G. Wiche’s lab, it was demonstrated that one such intermediary is plectin, particularly its 1b isoform, which interacts both with desmin and mitochondria [10]. However, experimental evidence showed certain desmin mutations which do not interfere with native IF structure or apparently with binding of plectin yet cause pathological conditions [11], including mitochondrial dysfunction [7]. A direct association between desmin IFs and mitochondria could explain these phenomena. In this study, we examined the possibility of a direct interaction between purified recombinant desmin and isolated rat liver mitochondria. We performed centrifugation in the sucrose gradient to separate desmin and a heavier mitochondrial fraction. To prevent sedimentation of unbound desmin, we used its mutant form Des(Y122L). This point mutation in desmin arrests its polymerization at the stage of unit-length filaments (ULFs) consisting of approximately 30–40 polypeptides of desmin that are much lighter than long IFs and sediment only at high-speed centrifugation. Figure 1 shows that Des(Y122L) when expressed in rat fibroblasts REF-52(Vim−/−) devoid of IFs is unable to form filaments. It rather forms ULFs evenly distributed throughout the cytoplasm, similarly to mutant vimentin(Y117L) [12,13].
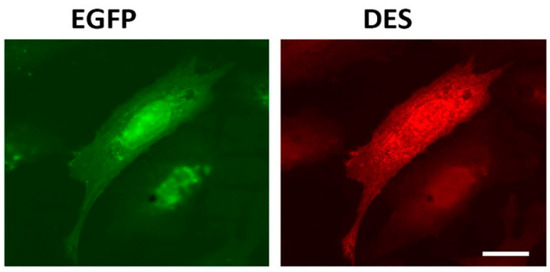
Figure 1. Immunofluorescence of REF-52(Vim−/−) cells transfected with plasmid pIRES-EGFP-Des(Y122L). Expression of EGFP (left) and desmin(Y122L) (right) in rat fibroblast stained with antibodies against desmin shows evenly distributed unit-length filaments (ULFs) but not long intermediate filament (IF). Scale 10 µm.
2. Results
Desmin Binds to Mitochondria In Vitro
Previously, we hypothesized [6] that, similarly to vimentin, an N-terminal part of desmin molecule could contain a targeting signal which localizes desmin to the outer mitochondrial membrane (OMM). Its moderately hydrophobic region spanning from threonine-17 to proline-36 is flanked by two groups of positively charged amino acids—a characteristic pattern of many proteins known to be localized to OMM [14]. To predict the presence of mitochondrial targeting peptide in the molecule of human desmin, we used the online program TargetP 1.1 [15] to analyze its sequence and found that such signal is present with a high probability (mTP—0.864). Therefore, according to the analysis of desmin’s primary structure, it belongs to the proteins predisposed to localize in mitochondria.
To test the possibility of the direct binding of desmin to mitochondria, we probed the interaction of mitochondria from a rat liver with a recombinant human Des(Y122L) in vitro. The protein was expressed and purified from bacterial lysate (see Supplementary Methods and Figure S1A). The bound Des(Y122L) was determined using Western blotting of mitochondrial pellets obtained by centrifugation of their mixtures through sucrose cushion.
When centrifuged through a sucrose cushion, such desmin mutant did not sediment by itself and completely stayed in the supernatant (Figure 2A,B, lines S1 and P1). However, when the sample contained mitochondria, some Des(Y122L) was found in the pellet, but only in the case where the protease inhibitor cocktail was present in the mixture (Figure 2A,B, lines S3 and P3). Incubation of this protein with mitochondria without protease inhibitors led to partial protein degradation, and the shortened Des(Y122L) was found only in the supernatant (Figure 2A,B, lines S2 and P2). COX-IV was used as a marker of mitochondrial proteins in the pellets and the loading control (Figure 2C). Hence, full-size Des(Y122L) bound to mitochondria and co-sedimented with them through sucrose cushion, while cleaved desmin did not.
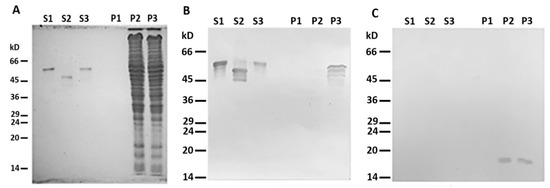
Figure 2. Desmin and mitochondria co-sedimentation. (A) SDS-PAGE of supernatants (S1, S2, S3) and pellets (P1, P2, P3) after centrifugation through sucrose cushion of mixtures of Des(Y122L) and mitochondria. Des(Y122L) was incubated without mitochondria (S1, P1), with mitochondria (S2, P2), or with mitochondria and protease inhibitor cocktail (S3, P3). Western blotting of the same samples as in (A) with antibodies against desmin or COX-IV is shown in (B,C), respectively. The desmin molecule approximately 10 kD smaller after the incubation with mitochondria (S2) does not co-sediment with mitochondria (P2), while the addition of protease inhibitor cocktail prevents desmin cleavage (S3) and ensures its co-sedimentation with mitochondria (P3). COX-IV is detectable only in lines P2 and P3 which shows the presence of mitochondrial proteins in the pellets.
More than 40 mitochondrial proteases are known to maintain normal functioning of these organelles [16]. We proposed that one (or several) of these mito-proteases may cleave Des(Y122L) when it interacts with mitochondria. To discover a candidate protease, we performed an inhibitory analysis. We examined several protease inhibitors, including PMSF (phenylmethylsulfonyl fluoride), aprotinin, TAME (Tosyl-L-Arginine Methyl Ester), leupeptin, and E-64, and found that only the last two inhibited desmin degradation (Figure 3). Since leupeptin and E-64 are known to inhibit cysteine proteases, in contrast to serine protease inhibitors PMSF, aprotinin, and TAME, we inferred that a candidate belongs to the cysteine proteases. Figure 3B shows that protection of Des(Y122L) against cysteine proteases allows its co-sedimentation with mitochondria.
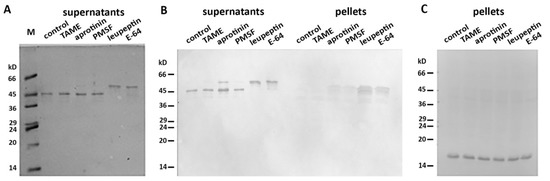
Figure 3. Inhibitory analysis of desmin degradation. Protease inhibitors are indicated. (A) Samples stained with Coomassie Brilliant Blue after electrophoresis in 15% SDS-PAGE of desmin and mitochondria mixtures after centrifugation through sucrose cushion. Only leupeptin and E-64 inhibitors prevent the formation of 45 kD desmin cleavage product. (B) Immunoblotting of desmin and mitochondria mixtures after centrifugation through sucrose cushion. Only intact desmin molecule co-sediments with mitochondria (leupeptin and E-64 lines in pellets). (C) Immunoblotting of COX-IV in the pellet fractions of the samples. COX-IV is detectable in all lines which show the presence of mitochondria.
To determine which type of cysteine protease was involved in desmin degradation, we used more specific inhibitors—calpeptin and PD150606. It turned out that calpeptin, which inhibits calpains and cathepsins L and K [17,18], effectively protected desmin (Figure 4, lines S1 and P1), while PD150606, which specifically inhibits calpains I and II [19], did not prevent protein degradation (Figure 4, lines S2 and P2). Since PD150606 inhibits only typical calpains that possess domain IV, we proposed that the protease responsible for desmin cleavage is atypical calpain-10, which has been found in mitochondria earlier [20]. Although the contamination of mitochondria with lysosomal cathepsins cannot be completely ruled out, their participation is unlikely since available data on degradation of desmin by cathepsin [21] show no clear electrophoretic fragments but rather smear-like pattern.
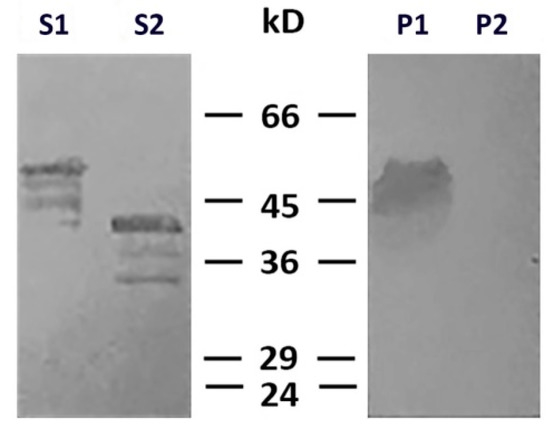
Figure 4. Effects of calpeptin and PD150606 on desmin and mitochondria co-sedimentation. Immunoblotting of desmin in supernatants (S1, S2) and pellets (P1, P2) after centrifugation through sucrose cushion. Desmin(Y122L) was incubated with mitochondria and calpeptin (S1, P1) or PD150606 (S2, P2). Non-cleaved desmin is seen both in supernatant and pellet only when calpeptin was added (S1, P1). Allosteric inhibitor PD150606 did not protect desmin from proteolysis (S2, P2).
N-terminus of Desmin is Necessary for Binding to Mitochondria
N- and C-termini of the desmin molecule are the most protease-sensitive regions, whereas the central alpha-helical domain is relatively stable [10,21]. Our data (Figures 2–4) show that the main product of desmin degradation resulting from the incubation with mitochondria was smaller than the initial polypeptide by approximately 10 kD. To identify which part of desmin molecule is cleaved, we performed mass-spectrometry analysis of the protein band in the gel corresponding to the cleavage product. The data demonstrated that desmin molecule was full-length when protected by leupeptin (Figure S1A) but lost its 70 amino acid-long fragment from N-terminus when incubated with mitochondria in the absence of inhibitors (Figure S1B). Hence, the cleavage of N-terminus might be responsible for the loss of ability of desmin to interact with mitochondria. Interestingly, the analysis of such shortened desmin using TargetP 1.1 showed very low probability of a mitochondrial target peptide in it (mTP—0.122).
To further elucidate the role of the N-terminal domain of desmin in its binding to mitochondria, we probed the interaction of the headless ΔN-Des(Y122L) obtained by the aforementioned procedure, including the incubation with mitochondria and subsequent centrifugation through sucrose gradient. We preferred such an approach to the expression of shortened polypeptide in bacteria because the formation of the dimers of type III IFs as well as the subsequent steps leading to generation of ULFs is strongly dependent on the intact N-terminal domain [22]. The analysis of the truncated ΔN-Des(Y122L) from the supernatants after centrifugation using SDS-PAGE showed that it was pure enough and in sufficient amount (Figures 2A and 3A). The data in Figure 5A demonstrate that ΔN-Des(Y122L) lacking N-terminus does not co-sediment with mitochondria in contrast to the entire non-truncated molecule that was treated similarly but in the presence of protease inhibitor leupeptin. Thus, the loss of N-terminal fragment of desmin molecule disrupts its binding to mitochondria.
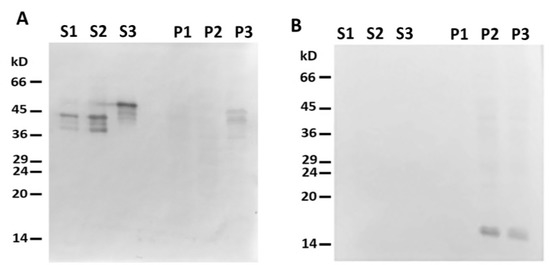
Figure 5. Headless ΔN-Des(Y122L) does not bind to mitochondria. (A) Western blotting of supernatants (S1, S2, S3) and pellets (P1, P2, P3) after centrifugation through sucrose cushion of mixtures of ΔN-Des(Y122L) (S2, P2) or Des(Y122L) (S3, P3) and mitochondria. The truncated protein ΔN-Des(Y122L) does not sediment without mitochondria (S1, P1). (B) Western blotting of the same samples as in A with antibodies against COX-IV shows the loading control of mitochondrial pellets.
To rule out the possibility of participation of the C-terminal domain in this interaction, we constructed the deletion mutant of desmin with the truncated C-terminal region 53 amino acids long. This ΔC-Des(Y122L) mutant also containing the point mutation Y122L was expressed in bacteria and purified similarly to the original protein (see Supplementary Methods and Figure S1B). Our data show that desmin missing C-terminus bound to mitochondria when its N-terminal part was protected from proteolysis by leupeptin (Figure 6, line P3). However, incubation in the absence of inhibitor led to its shortening and to the loss of the ability to co-sediment with mitochondria (Figure 5, lines S2 and P2). This protein did not sediment without mitochondria in these conditions (Figure 5, lines S1 and P1). The analysis of ΔC-Des(Y122L) by mass-spectrometry in the presence (Figure S1C) or in the absence of leupeptin (Figure S1D) showed that proteolysis led to the cleavage of the identical fragment (70 amino acids) to that of the whole Des(Y122L). Thus, N-terminal domain of desmin is necessary for the interaction with mitochondria, while C-terminus is unimportant.
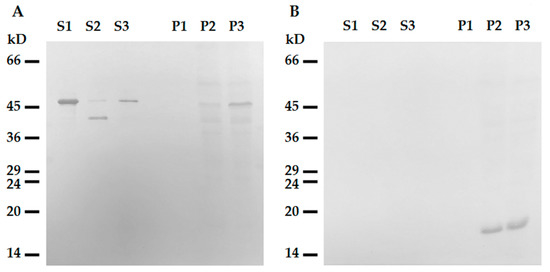
Figure 6. Interaction of tailless desmin and mitochondria. (A) Western blotting of ΔC-Des(Y122L) in supernatants (S1, S2, S3) and pellets (P1, P2, P3) after centrifugation through sucrose cushion. ΔC-Des(Y122L) was incubated 20 min without mitochondria (S1, P1), with mitochondria but without protease inhibitors (S2, P2), or with mitochondria and leupeptin (S3, P3). (B) Western blotting of COX-IV in the same samples shows loading control in mitochondrial pellets.
References
- Etienne-Manneville, S. Cytoplasmic intermediate filaments in cell biology. Rev. Cell Dev. Biol. 2018, 34, 11.1–11.28.
- Steen, K.; Chen, D.; Wang, F.; Majumdar, R.; Chen, S.; Kumar, S.; Lombard, D.B.; Weigert, R.; Zieman, A.G.; Parent, C.A.; et al. A role for keratins in supporting mitochondrial organization and function in skin keratinocytes. Biol. Cell 2020, doi:10.1091/mbc. E19-10-0565.
- Lehmann, S.M.; Leube, R.E.; Schwarz, N. Keratin 6a mutations lead to impaired mitochondrial quality control. J. Dermatol. 2020, 182, 636–647.
- Milner, D.J.; Mavroidis, M.; Weisleder, N.; Capetanaki, Y. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. Cell Biol. 2000, 150, 1283–1297.
- Capetanaki, Y. Desmin cytoskeleton: a potential regulator of muscle mitochondrial behavior and function. Trends Cardiovasc. Med. 2002, 12, 339–348.
- Chernoivanenko, I.S.; Matveeva, E.A.; Gelfand, V.I.; Goldman, R.D.; Minin, A.A. Mitochondrial membrane potential is regulated by vimentin intermediate filaments. FASEB J. 2015, 29, 820–827.
- Smolina, N.; Khudiakov, A.; Knyazeva, A.; Zlotina, A.; Sukhareva, K.; Kondratov, K.; Gogvadze, V.; Zhivotovsky, B.; Sejersen, T.; Kostareva, A. Desmin mutations result in mitochondrial dysfunction regardless of their aggregation properties. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165745.
- Fountoulakis, M.; Soumaka, E.; Rapti, K.; Mavroidis, M.; Tsangaris, G.; Maris, A.; Weisleder, N.; Capetanaki, Y. Alterations in the heart mitochondrial proteome in a desmin null heart failure model. Mol. Cell Cardiol. 2005, 38, 461–474.
- Cohen, S. Role of calpains in promoting desmin filaments depolymerization and muscle atrophy. BBA Cell Res. 2020, doi:10.1016/j.bbamcr.2020.118788.
- Winter, L.; Abrahamsberg, C.; Wiche, G. Plectin isoform 1b mediates mitochondrion-intermediate filament network linkage and controls organelle shape. Cell. Biol. 2008, 181, 903–911.
- Taylor, M.R.; Slavov, D.; Ku, L.; Di Lenarda, A.; Sinagra, G.; Carniel, E.; Haubold, K.; Boucek, M.M.; Ferguson, D.; Graw, S.L.; et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 2007, 115, 1244–1251, doi:10.1161/CIRCULATIONAHA.106.646778.
- Meier, M.; Padilla, G.P.; Herrmann, H.; Wedig, T.; Hergt, M.; Patel, T.R.; Burkhard, P. Vimentin coil 1A-A molecular switch involved in the initiation of filament elongation. Mol. Biol. 2009, 390, 245–261.
- Nekrasova, O.E.; Mendez, M.G.; Chernoivanenko, I.S.; Tyurin-Kuzmin, P.A.; Kuczmarski, E.R.; Gelfand, V.I.; Goldman, R.D.; Minin, A.A. Vimentin intermediate filaments modulate the motility of mitochondria. Biol. Cell 2011, 22, 2282–2289.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21218122

