

The hazardous effects of pollutants in the atmosphere are an increasing problem. Measuring the impact of these pollutants on human health remains challenging as only a direct risk assessment can be done by human biomonitoring. To tackle this hurdle, biologists and toxicologists rely on computational modeling assessing the impact of these compounds on public health. Physiologically-based pharmacokinetic (PBPK)models allow reproducing the toxicokinetics (TK) and toxicodynamics (TD) of these compounds. These models exist of different compartments, each of them representing a specific organ of the human body. This manuscript describes the application of these models to evaluate the impact of hazardous pollutants on human health. More in particular, the application of these models towards polychlorinated biphenyls (PCBs) will be discussed. The strengths and limits of PBPK modeling are mentioned as well. An opinion on applying PBPK modeling of PCBs for regulatory purposes is provided. It is shown that these models can support policies of regulatory authorities based on risk assessments of PCBs and even other pollutants.
- Hazardous pollutants
- PCB
- polychlorinated biphenyls
- PBPK
- physiologically-based pharmacokinetic modeling
- toxicology
Introduction
The presence of polychlorinated biphenyls (PCBs) in the environment is an impacting problem. The risk for human health should not be underestimated and policy guidelines are targeted at reducing human exposure. In general, measuring the impact of persistent organic pollutants (POP), of which some are carcinogenic and mutagenic, has evidenced their hazardous effects on human health. We recently reviewed dioxin incidents and - more in particular - polychlorinated biphenyl (PCB) incidents, throughout the 20th century (Hens et al., 2016; Hens and Hens, 2017). The potential for these molecules to interact with DNA and causing damage via activation of the nuclear aryl hydrocarbon (AhR) receptor was demonstrated by cross-sectional relationship studies and in in vitro/in vivo in laboratory animals to confirm the hazardous effects on the DNA transcription/translation process (Aoki, 2001; Bock, 2013; Faroon and Ruiz, 2015). However, the idealized exposure as created in in vitro experiments might underestimate the “real-life” situation, as humans are exposed to a ‘cocktail’ of many pollutants in daily life (the entire PCB family consists of 209 congeners). The interplay between human biomonitoring (pharmacokinetics) and in vitro/ in vivo studies to evaluate the hazardous effects (pharmacodynamics) is of utmost importance to support policy guidelines on thresholds of maximal exposure levels to safeguard human health. Nevertheless, one should be careful in drawing conclusions between systemic exposure to the pollutant and the toxicological effects. The link between the exposure and the adverse effect is not always linear. Moreover, extrapolation from animals to humans should be taken cautiously as there are major differences in anatomy and physiology between different species (Li et al., 2018). Depending on the physicochemical properties and the tissue/blood partition of the pollutant, the balance between accumulation of the compound in the body versus the clearance of the molecule from the body can be extremely variable. Although human biomonitoring helps us directly quantifying the exposure to a pollutant in the human body as measured from plasma samples, latent effects of pollutant exposure may only be visible after a latency period as these molecules are characterized by lipophilic properties, accumulating in the lipophilic tissues. In addition, human biomonitoring induces ethical considerations which hamper human ecological research.There are uncertainties related to the ‘real-life’ impact of hazardous pollutants such as PCBs on human health. To overcome this, a lot of attention goes out to physiologically-based pharmacokinetic (PBPK) modeling. PBPK modeling reflects human/animal anatomy and physiology using different compartments, each of them reflecting a specific organ of the human body. Each organ is regulated using differential mass-balance equations to simulate the in-house dynamic interactions and householding of the tissue (e.g., arterial/venous blood flow). The purpose of this model is to describe drug concentrations in various tissues (e.g., plasma, liver, kidney) as a function of time. Based on measured concentrations of pollutants in the environment, the PBPK model can be fed with this information in order to extract and predict the exposure for an individual. The history of PBPK modeling goes back to 1937, when the Swedish physiologist Torsten Teorell published two papers describing the circulatory system, a drug depot, fluid volume, kidney elimination and tissue inactivation as five different compartments (Rescigno, 1997). More insights into physiology came twenty years later when Edelman and Liebman developed a more sophisticated model dividing total body water into plasma, interstitial lymph, inaccessible bone water, and transcellular versus intracellular components (Edelman and Leibman, 1959). In 1968, Bischoff and Dedrick succeeded in reproducing the pharmacokinetic disposition of thiopental and methotrexate (Bischoff et al., 1971; Bischoff and Dedrick, 1968). Over the years, more knowledge was gained with respect to the physiology of the human body, resulting in better predictions and the first reviews related to PBPK models used for certain classes of drug compounds. In 1981, John Wagner focused on population covariates to optimize dose regimens for each individual (Wagner, 1981). The inclusion of specific covariates opened horizons to obtain population estimates of pharmacokinetic parameters and their variabilities, therapeutic monitoring and prediction of the time course of the intensity of pharmacological effects, which has now been turned into reality in the commercially available PBPK software programs (Kostewicz et al., 2014).
This review provides an overview of the applications of PBPK modeling to predict the exposure of hazardous chemicals such as PCBs in humans/animals. First, the different pharmacokinetic models that are applied to simulate the absorption, distribution, metabolism and excretion (ADME) properties of a chemical compound are discussed. The importance of this strategy to map the concentrations of PCB molecules in the organs is considered. The strengths and limits of PBPK modeling are mentioned. Finally, the application of PBPK modeling of PCBs in a regulatory context will be discussed with contemporary examples.
Materials and methods: literature review
Papers published in peer-reviewed journals were searched in the ‘Pubmed’ database of the US National Library of Medicine National Institutes of Health and selected on the keywords: polychlorinated biphenyls OR modeling OR PBPK modeling OR pharmacokinetic modeling OR PK analysis OR compartmental modeling. As a first screen, only articles from 2000 on were considered. The next selection step was based on the apparent relevance of the title. After reading the abstract, a final selection was made. Articles published before 2000 were selected based on the snowball sampling technique. This resulted in a core number of 110 papers published in international, peer-reviewed journals, on which this review is based.
Physiologically-based pharmacokinetic (PBPK) models: how and where to start?
‘Pharmacokinetics’ means ‘the kinetics of the pharmakon’, the Greek word for drugs and poisons (Wagner, 1981). The main goal of pharmacokinetic modeling is to describe the concentrations of a chemical as a function of time in any tissue of interest in the body. From that point of view, pharmacokinetic models take into account all facets influencing the absorption, distribution, metabolism and excretion (ADME) of compounds (Rowland et al., 2011b, 2011a). Historically, these models were developed to describe drug concentrations in the human/animal body after administration. Therefore, the predicted outcomes were able to support or reject the new drug candidates with a high potential to reach the market because of their therapeutic/pharmacodynamic effects. In a preclinical phase of drug development, these models are frequently applied by pharmaceutical companies to provide relevant information on the therapeutic indications and/or the toxic/adverse events (i.e., physiologically-based toxicokinetic (PBTK) modeling) that can be expected later on during the clinical trials (Pearce et al., 2017). Even more, predicted data derived from pharmacokinetic models can be used to support the approval of a new drug (NDA) (Zhao et al., 2011).
Overall, three basic types of pharmacokinetic models exist: (i) compartmental models, (ii) physiologically-based pharmacokinetic (PBPK) models and (iii) non-compartmental models, which use time and drug concentration averages (Loftsson, 2015). Related to the first type, compartmental models consider different tissues (all represented as single compartments). In every compartment, the drug is homogeneously mixed. Transfer from one compartment to another occurs by simple first-order rate equations (i.e., a constant fraction of the drug will be transported to another compartment) or zero-order rate equations (i.e., a constant amount of drug will be transported to another compartment). Establishing compartmental models can be done by using available software programs such as WinNonlin®, Stella® and Berkeley Madonna®. These programs allow starting from scratch and draw as many compartments as intended by the purposes. A central compartment connected to peripheral compartments (e.g., fat tissue) is one of the most common – and most simple - models to construct describing ADME properties of simple molecules (Figure 1).
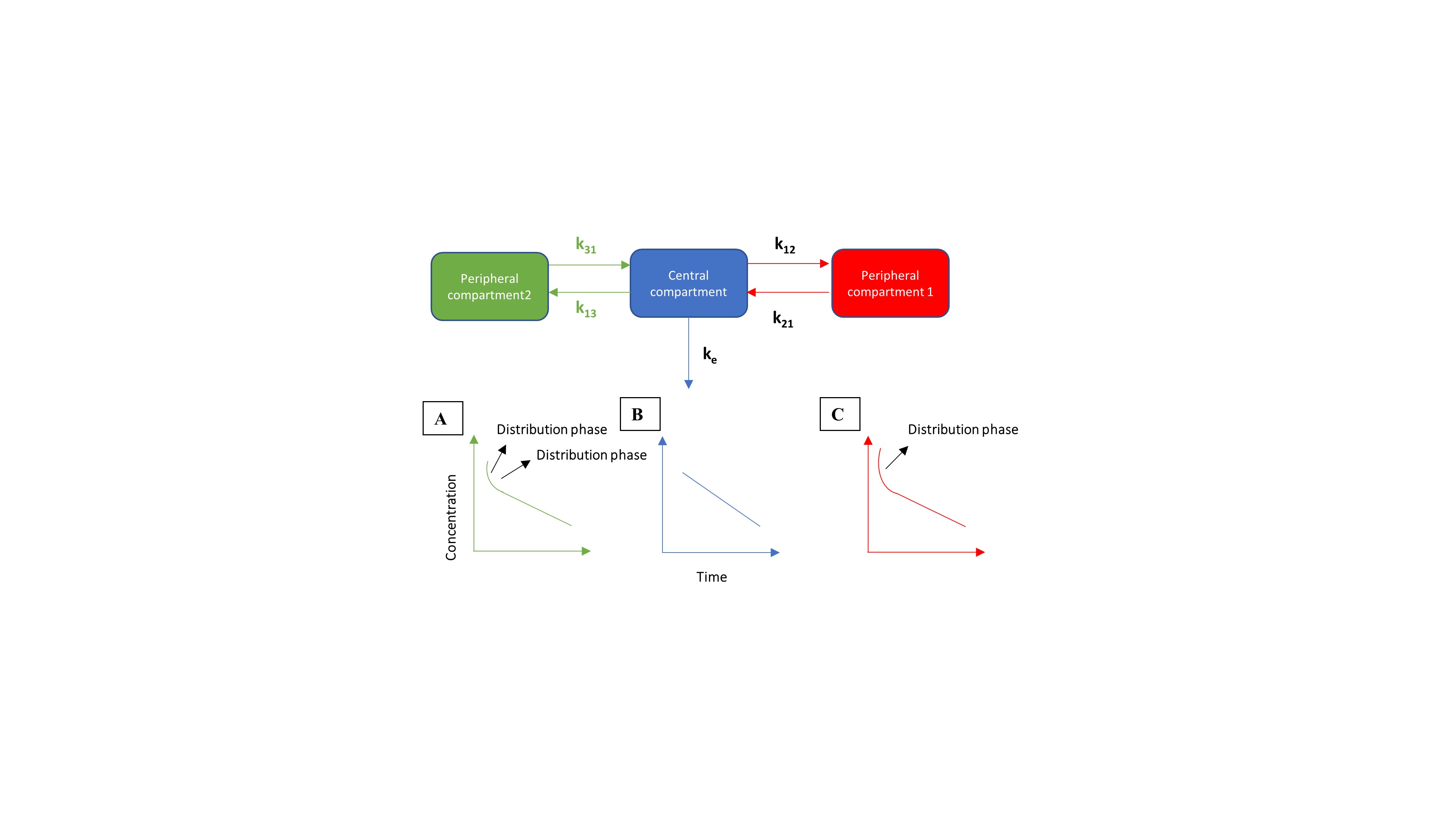
Figure 1: Example of a one (blue), a two (red) and a three (green) compartmental model. After intravenous dosing in the central compartment, the molecule will be distributed between the central and peripheral compartment(s), regulated by the distribution rate constants k12, k21, k13 and k31. Clearance of the compound is regulated by the elimination rate constant ke. (A) Simulated concentration-time profile for a three-compartmental model, consisting of two distribution phases. (B) Simulated concentration-time profile for a one-compartmental model. (C) Simulated concentration-time profile for a two-compartmental model, consisting of one distribution phase.
A simple, one-compartmental model (only containing a central compartment), assumes that changes in systemic concentrations are proportional to changes in peripheral concentrations. The elimination rate constant (ke) determines the slope of the concentration curve. After exposure to PCB molecules, distribution of the chemical can be observed among multiple organs (e.g., liver, kidneys, lungs). In this situation, a more multi-compartmental approach is needed to adequately describe drug distribution and disposition along the human body. Two components to simulate the redistribution are necessary to highliht the slow elimination process and bioaccumulation of lippophilic compounds in fatty tissue. This was observed for PCB 153 where k21 was so much lower than k12 that elimination could not be determined (Hansen, 2012). To have an accurate simulation of that specific situation, a multi-compartmental approach is more appropriate (Figure 1). Linking a central compartment with a peripheral compartment allows to reproduce systemic concentrations and concentrations in fat tissues. In case of lipophilic compounds, such as PCBs, simulated peripheral concentrations can be compared with observed data, whenever available. To model the more complex situation of human/animal physiology and anatomy, full PBPK models more adequately describe the distribution of a compound over all organs of the body (Kostewicz et al., 2014). Each compartment in this model is closely related to the anatomy and physiology of a specific organ with respect to its volume and chemical binding (i.e., tissue to blood partitioning (kp)). These models integrate the blood flow (Q) for each organ, which is obviously necessary to produce tissue concentrations of the compound of interest (Figure 2).
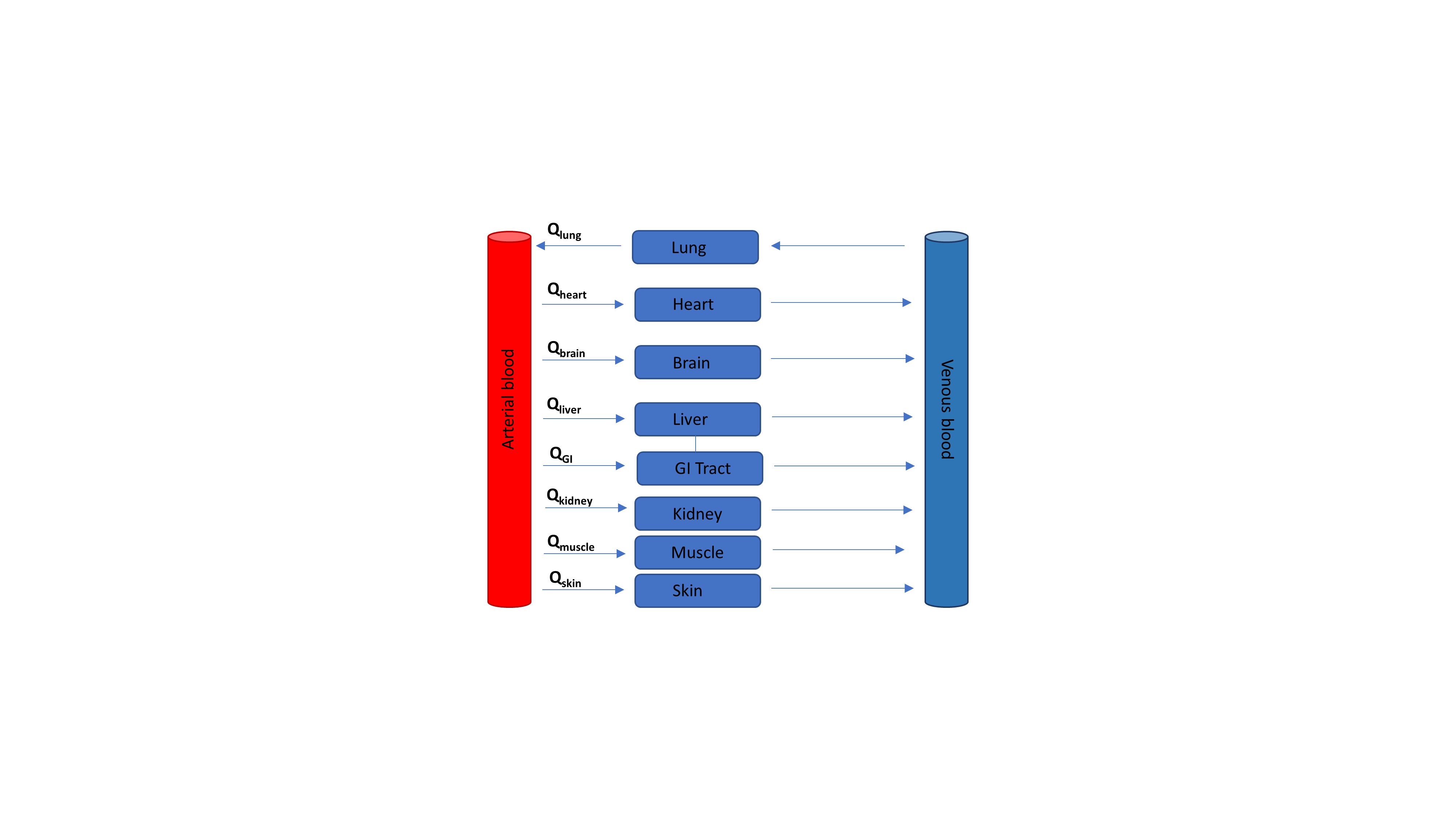
Figure 2: Illustration of a physiologically-based pharmacokinetic model. ‘Q’ represents the blood flow in each specific organ. ‘GI’ stands for ‘gastrointestinal.
This implies that more information and knowledge is needed to rely on the outcomes predicted by these models. Nowadays, commercially-available software programs (e.g., GastroPlus™, Simcyp®) are available that assist in predicting the systemic and tissue concentrations of the compound of interest by feeding the program with physicochemical (e.g., LogP, pKa) and biopharmaceutical (e.g., route of administration, dosage form) information. These characteristics of the compound are important to adequately describe the drug distribution over the various tissues of the PBPK model. The first approach of PBPK modeling for drug compounds was performed by Teorell and co-workers in 1937 (Teorell, 1937a, 1937b). This work applied a five-compartmental model representing the circulatory system, a drug depot, fluid volume, kidney elimination and tissue inactivation. The work stimulated other researchers to develop more advanced PBPK models which are more ‘biorelevant’. The use of PBPK modeling for volatile anesthetics was first described in 1924 by Haggard who focused on the implementation of the role of ventilation, blood flow and blood versus air solubilities in chemical uptake in the body (Haggard, 1924). More specific work related to refining these models was done in the seventies (Chen and Gross, 1979). One of the major drawbacks in 1920-1930s was the limited knowledge on human/animal anatomy and physiology as well as the computational skills to model all these parameters in one program. However, initiatives and efforts paid off as PBPK modeling is increasing in a number of peer-reviews publications during the last decades (Figure 3).
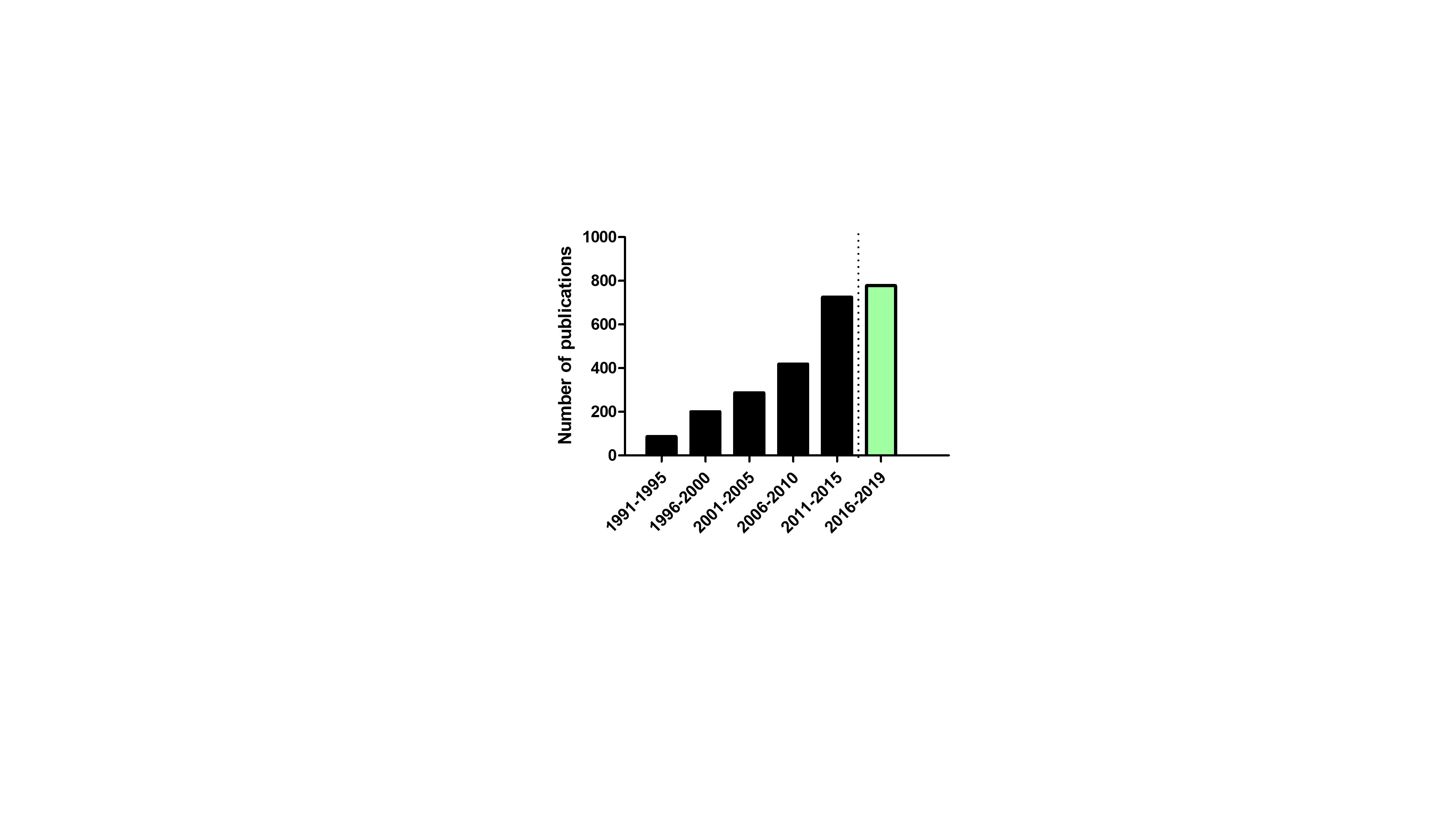
Figure 3: Number of publications per five years in the ISI Web of Knowledge® database mapping to the topics ‘PBPK Modeling’. The green bar represents the number of publication from 2016 until to date (last accessed on 01/08/2019).
As both previous models are able to describe concentrations as a function of time by applying kinetic models, non-compartmental models estimate the exposure of a drug by integrating the area under the curve (AUC) of a drug-concentration time profile applying the trapezoidal method. In addition, pharmacokinetic parameters such as plasma Tmax, Cmax, half-life ( ), distribution coefficients and clearance can be calculated. The advantage of non-compartmental models is that these models can predict drug and disposition parameters in a high-througput way. Although these ‘static’ models are very useful for predictions of overall drug exposures in humans (in terms of AUC), they rely on steady-state assumptions and, therefore, are not able to predict the plasma concentration- curve as a function of time, nor time-varying changes in enzyme or transporter inhibition (Sager et al., 2015). Moreover, the major drawback is that these calculations are estimations (i.e., steady-state assumptions), showing a lack of accuracy and precision (Loftsson, 2015).
Population pharmacokinetics
It is not unusual to observe considerable differences among people in exposure after contact with hazardous pollutants. This ‘inter-individual’ variability is caused by intersubject intrinsic and extrinsic factors such as environmental parameters, gender, genetic differences (i.e., expression levels of proteins), chemical interactions, age and body weight (Berggren et al., 2007; Canaparo et al., 2007; Englund et al., 2006). These covariates are responsible factors altering systemic exposure in such way that some subjects of a population could be more vulnerable after being exposed to certain pollutants than others due to genetic differences (e.g., different expression levels of metabolizing enzymes). Population pharmacokinetic modeling seeks to identify these pathophysiological covariates that alter the systemic outcome of a chemical after exposure (Loftsson, 2015). From a pharmacodynamic point of view, these models can be applied to adjust doses from person to person to reach the targeted therapeutic outcome for each individual. With respect to environmental ecology, these models can assist scientists to find vulnerable persons for the pollutants. This allows reducing potential risks and preventing adverse effects for a population. Sharma and co-workers developed a pregnancy-PBPK (P-PBPK) model to predict the exposure of bisphenol A (BPA) for the fetus based on the mother’s exposure (Sharma et al., 2018). Therefore, the authors extended an adult PBPK model by adding a placenta and fetus sub-compartment. Both the adult PBPK and P-PBPK model were validated with clinical data from human biomonitoring studies. Ongoing validation and optimization of this transplacental model are necessary to better understand the exposure to toxic chemicals (Binnington et al., 2016; Lee et al., 2002; Wood et al., 2016). Another study from Ulalszewska and co-workers defined a PBPK model to address PCB levels in breast milk of Italian women (Ulaszewska et al., 2012). This work presents a PBPK model of a woman in connection with a PBPK model describing the physiology of a fetus. The predicted outcomes were compared with observed data to validate the model. Moreover, published toxic tresholds of PCB concentrations (expressed as toxic equivalent factors (TEFs) and toxic equivalents (TEQs)) can be applied to evaluate the risk for mother and fetus (Van den Berg et al., 1998).
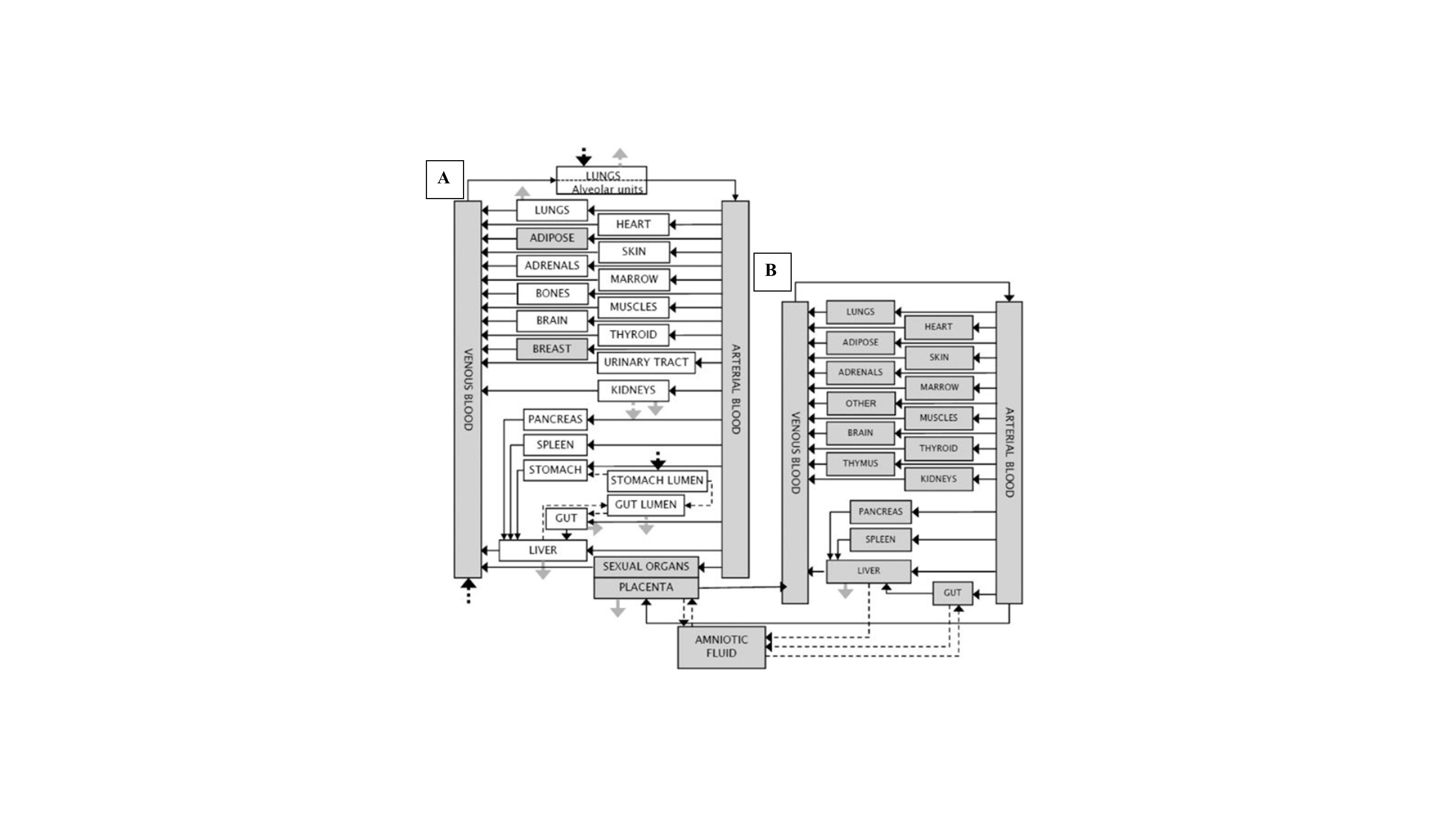
Figure 4: Schematic representation of the general PBPK model for a (A)woman and a (B) fetus. The compartments evolving during pregnancy and lactation, and the fetal submodel are in grey. Connections between compartments via blood flow are represented with this dark arrows, and the connections via diffusion processes with thin dotted dark arrows. The thick dotted dark arrows are the possible administration routes (i.e., via lungs, stomach lumen and venous blood). Metabolism is represented in plain grey arrows and the potential excretion routes in dotted grey arrows. Adapted from Ulalszewska and co-workers (Ulaszewska et al., 2012). Copyright Nature Publishing Group 2012.
Overview of available PBPK models
Toxicokinetic modeling of hazardous compounds is a valuable tool to explore the toxic mechanisms of drugs after administration. These models allow not only to predict systemic concentrations, but also tissue concentrations. Furthermore, commercially available software packages are able to easily scale-up from animals to human. Although these models have the same goal, there are some structural differences which will be discussed in this section.
The Simcyp Simulator®
The Advanced Dissolution Absorption and Metabolism (ADAM) is a multi-compartmental gastrointestinal transit model fully integrated into the Simcyp Simulator® (Certara, Princeton, NJ) as well as the rat, mouse and dog simulators. The Simulator provides the physiology of a Caucasian population and a Japanese population but also focuses on patient populations such as cancer patients (Jamei et al., 2009). The ADAM model consists of 9 compartments, representing the entire gastrointestinal tract. Mathematical equations to describe disintegration, dissolution and absorption of the chemical are integrated in the programming code. Moreover, based on literature data, the model includes inter-subject variability related to the pH and bile salt concentrations. Fluid volumes are handled as a continuous, dynamic process based on MRI work that explored the movement of residual fluids in the human gastrointestinal tract as a function of time in fasted and fed state. Chemicals entering the enterocytes can be metabolized by gut wall metabolic enzymes. Also, intestinal uptake/efflux transporters are present in these cells.
GastroPlus™
GastroPlus™ (Simulations Plus, Lancaster, CA) applies the advanced compartmental absorption transit (ACAT) model to represent the human gastrointestinal tract, provided with mathematical equations to describe the different intraluminal processes of drug release, dissolution and absorption (Agoram et al., 2001; Lukacova et al., 2009). This model contains 9 compartments, covering the entire gastrointestinal tract. The simulator applies different modules that consider physiology of different populations (e.g., American (Western) and Japanese (Asian) population). The PBPKPlus™ module is able to calculate tissue:plasma partition coefficients (Kp) or convenient import of user-specified Kp values. The physiology of beagle dogs, rats, monkeys, and mice are also available for modeling preclinical in vivo data. Chemicals can be metabolized and transported by enzymes and transporters, respectively. Most of the equations involve linear kinetics, whilst Michaelis–Menten nonlinear kinetics is used to describing saturable metabolism and carrier-mediated transport.
PK-Sim®
In the current version of PK-Sim®, 12 compartments represent the human gastrointestinal tract (Willmann et al., 2012). All other organs (e.g., liver, kidneys) are presented as compartments and each compartment is subdivided into a vascular and an extravascular space. Further, the vascular is subdivided into red blood cells and plasma, whereas the extravascular space consists of the interstitium and the cellular space. The compartments contain biorelevant concentrations of lipids, proteins and water to adequately reflect the tissue:plasma partition coefficient (Kp). Similar to the other models, PK-Sim® handles first pass metabolism using various kinetic models (e.g., Michaelis–Menten or first-order kinetics). For this purpose, the corresponding enzyme abundance for each organ (such as the liver or the various GI compartments) can be incorporated into the software.
User-customized models: Berkeley-Madonna®, Matlab®, Stella® and WinNonlin®
In addition to commercially-available models, user-customized models are interesting platforms to build your own model from scratch. These customized models such as Berkeley-Madonna®, Matlab®, WinNonlin® and Stella® allow to perform simulations for multiple purposes beyond generic PBPK, as for instance PBPK-PD or modeling of in vitro transporter kinetics and other cellular process. Kinetics between the different compartments can be described by mass-balance first/zero order equations. An illustrative example together with associated mass balance equations is given in Figure 5.
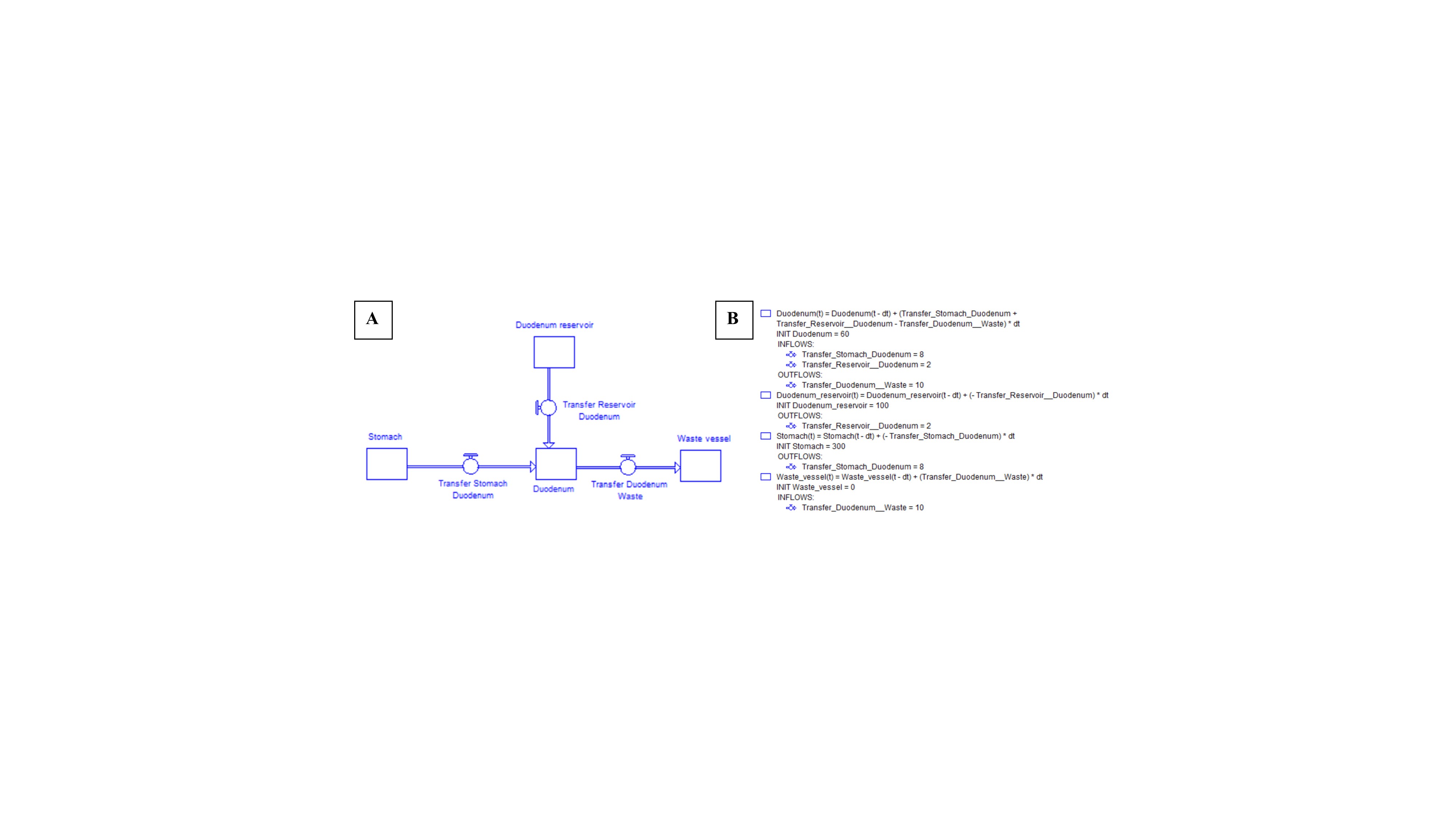
Figure 5: (A) Example of a four-compartmental model simulating the upper gastrointestinal tract. (B) Mathematical equations describing the mass transport of the drug between the compartments.
Application of PBPK modeling in toxicological risk assessment
Toxicological risk assessment focuses on four different topics: (i) hazard identification, (ii) dose-response relationships, (iii) exposure assessment and (iv) risk characterization (Andersen, 1995; Andersen et al., 1987; Cogliano, 1998; Larsen, 2006; Lind et al., 2004; Masuda, 1996; Mills and Andersen, 1993). Because dose-response relationships are not always linear – for instance, for endocrine disrupting agents or due to saturable uptake/efflux transporters in the gastrointestinal tract – researchers rely on PBPK modeling as these models can uncover the underlying mechanisms and, therefore, improving dose-response relationships. Nowadays, animal toxicity studies in combination with in vitro experiments are performed to define the levels of toxicity and safety (Figure 6) (Pizzo and Benfenati, 2016).
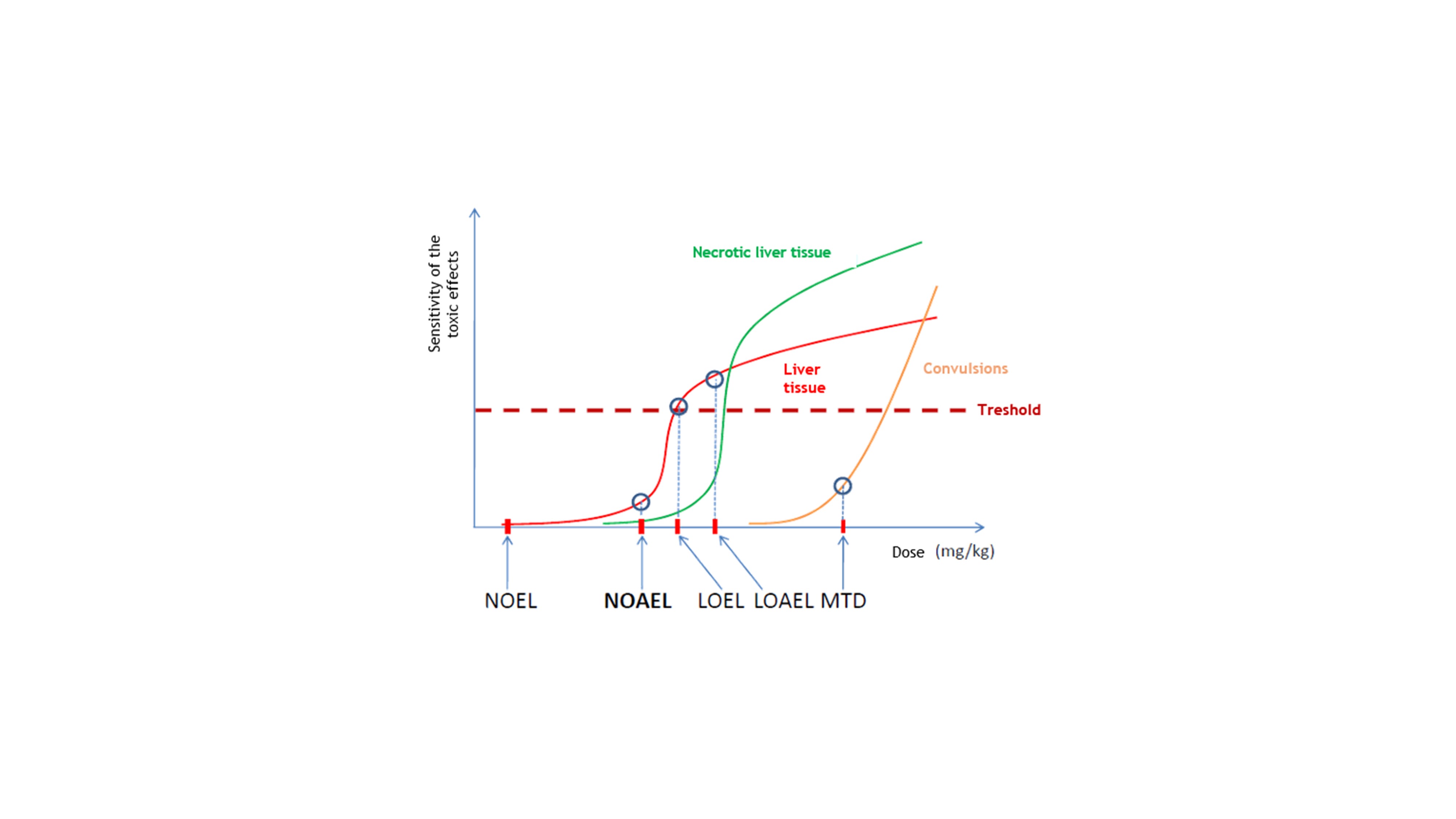
Figure 6: representative illustration of the toxic effects that may occur after dose escalation. NOEL: no observable effect level; NOAEL: no observable adverse effect level; LOEL: lowest observable effect level; LOAEL: lowest observable adverse effect level; MTD: maximum tolerated dose. Figure based on Figure 14 from Moffett and co-workers (Moffett et al., 2007).
In the end, and as wisely mentioned by Paracelsus (1493-1541): “All substances are poison: there is none which is not a poison. The dose differentiates a poison and a remedy.” The information generated by (i) dose-escalation studies in animals and (ii) interaction studies performed in vitro are a valuable input for PBPK modeling to predict toxic thresholds in humans after exposure to the chemical/pollutant of interest (i.e., physiologically-based toxicokinetic (PBTK) modeling). This is related to the required information needed to adequately build a PBPK model. First, physiological and anatomical knowledge should be correctly organized in the model fully respecting the human/animal physiology. Second, biochemical and physicochemical parameters such as the metabolizing rate and tissue-partition coefficients are of utmost importance to reflect elimination and tissue distribution. At last, the PBPK model should rely on a precise route of administration and timing of the simulation (acute versus chronic exposure). It is of paramount importance to keep these three pillars in mind (Figure 7) (Andersen, 1995).

Figure 7: Representative illustration of the important aspects that should be considered in order to develop an accurate and adequate PBPK model: (i) implementation of physiological variables and processes, (ii) information with respect to biochemistry, physiology and blood:tissue partitioning of the compound and (iii) the acquired route of administration and simulation enduration. Copyright Elsevier 1995. Figure derived from Andersen, 1995 (Andersen, 1995).
To validate the PBPK model, the predicted outcomes should be compared with the reference data from human biomonitoring studies, if available. Validation and, if necessary, optimization are essential to fully rely on these models in the future. A post-hoc sensitivity analysis test can be performed as a quantitative assessment of uncertainty to characterize the confidence bounds in a model output (Ozkaynak et al., 2008). As highlighted in Figure 7, gathering physicochemical data are an indispensable input for a PBPK model to rely on confidential predictions. Tissue partition coefficients are valuable parameters that describe the concentration ratio of two adjacent compartments at equilibrium (e.g., Schmitt’s model) (Peyret et al., 2010; Schmitt, 2008). Although many physicochemical/biological parameters can be determined by in vitro/ in vivo experiments, there is a major interest to predict these values by relatively high throughput toxicokinetic (HTTK) methods (Cohen Hubal et al., 2019; Pearce et al., 2017). These methods are able to predict the different properties of a compound by quantitative structure-activity relationships (QSAR) (Ruiz et al., 2014; Zhu et al., 2013). Based on a series of compounds with known physicochemical properties, predictions towards new chemical entities’ properties can be done. The HTTK methods are applied by the pharmaceutical companies to determine the range of efficacious doses in a preclinical stage of drug development (Jamei et al., 2009; Lukacova et al., 2009). These HTTK methods are illustrated in software packages such as Simcyp® and GastroPlus™. Extrapolation of in vitro data to in vivo exposure as developed for pharmaceuticals can easily be adapted to chemicals in the environment. A first example is the model of Rotroff and colleagues who predicted blood concentrations of chemicals after chronic daily exposure (Rotroff et al., 2010). Their model was validated with experimental data. A simple compartmental model was built with the assumption that the predicted plasma concentrations were equal to the peripheral concentrations in the tissues. In addition, extrahepatic metabolism was neglected and the model assumed 100% oral absorption after oral administration. In a next step, researchers aimed to expand the model towards population modeling (Sipes et al., 2017). Therefore, differences in physiology, genetics, and disease state can lead to different internal chemical concentrations following external exposure to a chemical. To simulate different scenarios with all possible outcomes for a specific population, Monte Carlo simulations can be performed (Morin, 2017). Numerous papers have been published by several authors in recent years on this topic, in particular to assess the impact of variability and uncertainty on the estimates of internal dose for different chemical exposures contexts (Sasso et al., 2012). These approaches use 'prior' information of the parameters, either from the literature or from previous model runs. The advantage is that this method uses such 'prior' parameters to calculate probability distributions to determine 'posterior' values that best explain the field observations among a population of any species. Weijs and colleagues applied a Bayesian approach executed with Markov chain Monte Carlo (MCMC) simulations to explain the observed bioaccumulation and pharmacokinetics of PCB 153 in long-finned pilot whales (Weijs et al., 2014).
Toxicokinetics (TK) versus toxicodynamics (TD): linear or not linear?
Dose-response relationships are not always linear and should be fully explored to unravel the toxic pathway of hazardous pollutants and their impact on human health. Nonlinear effect models can be applied to explain the toxicodynamic (TD) effects observed after titration of different doses of toxic markers such as PCBs. Dichloromethane was one of the first examples to demonstrate nonlinear effects after being exposed to different doses of the molecule. Investigating the interaction with the metabolic enzymes was important to fully understand its disposition.
With respect to PCBs, a nonlinear correlation was observed between dosing of PCB126 and the hormone levels present in the hypothalamic-pituitary-thyroid (HPT) axis in the rat. After oral administration of different PCB126 doses to Sprague-Dawley rats, induction of hepatic UDPGTs by the aryl hydrocarbon receptor AhR were noticed. However, the dose-response characteristics of the HPT axis were nonlinear and complex, requiring sophisticated tools, such as PBPK models, to characterize dose response (Fisher et al., 2006). With respect to PCB 197, 202, 194 and 95, a nonlinear correlation was observed when increasing amounts of these congeners were tested for activity towards ryanodine-sensitive Ca2+ channels (RyR). These channels are associated with neurotoxicity in exposed animals. RyR-active congeners follow a distinct structure–activity relationship and, based on quantitative structure–activity relationship (QSAR), it is predicted that a large number of PCBs likely activate the receptor (Holland et al., 2017). Another independent study of Sasso and co-workers demonstrated the need of incorporating a mass balance of CYP1A and CYP2B to adequately predict PCB metabolism at high doses (Sasso et al., 2012).
At the National Academy of Sciences Meeting in 1986, biologists and toxicologists debated this more mechanistic approach establishing risk assessment for hazardous chemicals. Later on, the following paragraph was published in a summary report of that meeting (National Research Council, 2001): “…PBPK analysis has genuine promise for improving risk assessment. We shall explore the strengths and weaknesses of PBPK analysis, evaluate how such analysis may improve the process of risk assessment, and identify the areas of research that will lead to further improvement of our procedures and concepts.”
PBPK modeling of PCB molecules and derivatives
Among the most important groups of pollutants with hazardous effects on human health are benzene-derivatives, including mono- and non-ortho PCBs. Human exposure to PCBs mostly occurs by the oral route (throughout food contamination of the food chain). In human cells, PCBs disrupt/interfere the signal transduction pathway of a receptor resulting in the differentiation of tissue, cellular and biochemical processes. After diffusion in the nuclear membrane, it binds to the AhR receptor, as mentioned in the introduction (Bock, 2013; Brennan et al., 2015). Subsequently, this activated complex binds to the transcription factor AhR and the new complex ‘agonist-AhR-Arnt’ enters the nucleus of the cell and influences the expression of human genes by interfering with their promoter sequences (Hens and Hens, 2017; Kohn et al., 1993; Mills and Andersen, 1993; Parham et al., 1997). One of these genes is responsible for the expression of CYP1A2. Quantitative PBPK/PD models for PCBs must express this induction in reasonable biological detail and in mathematical equations that allow predicting the extent of induction at a range of dioxin doses and in various species (Andersen, 1995). Multiple models, varying from one to more compartments, have been developed to predict the dose-effect relationship for human exposure to different doses of dioxins (Andersen et al., 1993; Kedderis et al., 1993; Kohn et al., 1993). The CYP1A2 metabolism in these models is not linked with messenger RNA transcription, mRNA processing or translation. These models apply rate and binding constants (to reflect gene induction), which are curve-fitted based on human biomonitoring data (e.g., plasma). It would be indicated unraveling the process of transcription and translation of these proteins in order to have a representative reflection of these mechanisms of action in existing PBPK/PD models, especially at different doses. This remains extremely challenging. One of these models that has intensively been used and optimized is the concentration and age-dependent model (CADM). It was first developed by Carrier et al. and later on refined by Aylward and colleagues (Aylward et al., 2005; Carrier et al., 1995). More recently, Ruiz and co-workers re-coded the PBPK program into Berkeley-Madonna software (Ruiz et al., 2014) (Figure 8).
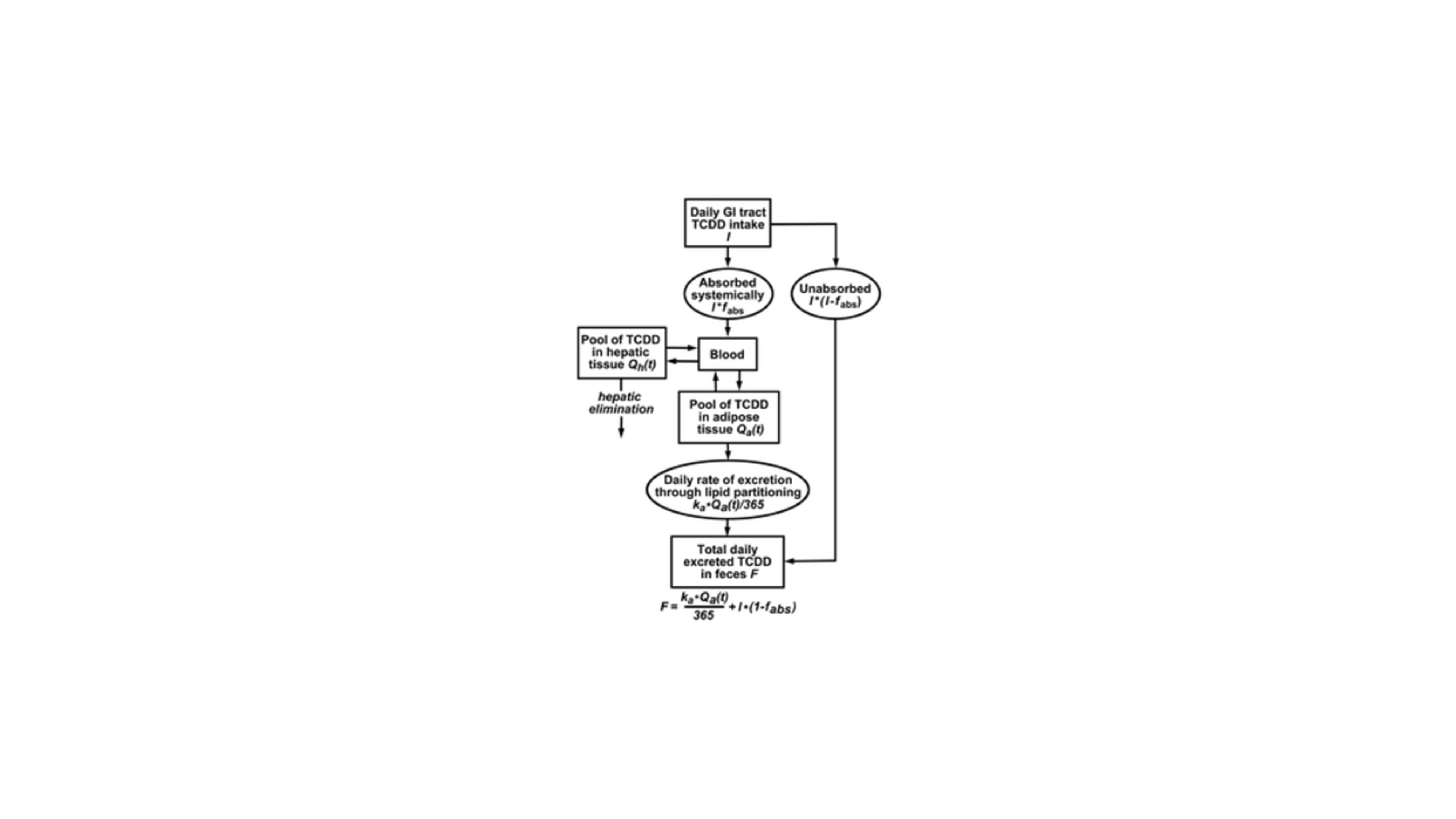
Figure 8: Illustration of the PCB model applied by Ruiz and colleagues to simulate PCB concentrations for different doses in human subjects. It should be noted that the authors optimized this model to reflect the induction of CYP1A2 enzymes after exposure of PCB molecules. Copyright Taylor & Francis 2014. Figure from Ruiz and co-workers, 2014 (Ruiz et al., 2014).
Validation of the recoded model was based on several clinical data sets: evaluation data from the case of Victor Yushchenko (former president of Ukraine), who was exposed to high concentrations of chlorine-containing hydrocarbons such as PCB, were used to simulate serum lipid TCDD. Human biomonitoring data of tissue concentrations (liver and adipose tissue) were provided by the literature and were used as a reference to validate the modeling results (Sorg et al., 2009). Simulated and observed results are provided in Figure 9.
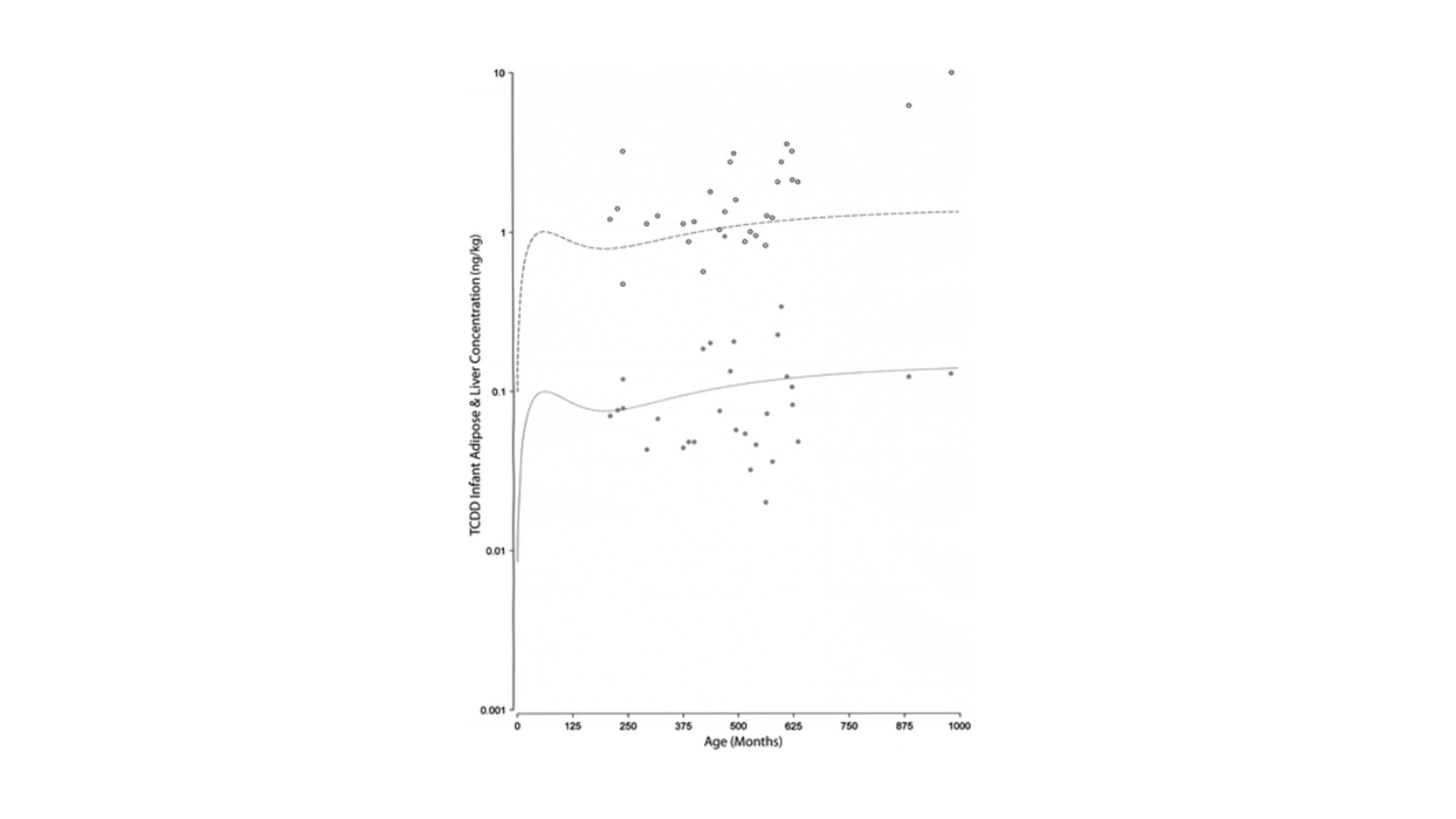
Figure 9: Concentration of TCDD in liver (grey dots) and adipose tissue (black open circles) as a function of time (poisoning data from Viktor Yushenko, former president of Ukraine). The lines show model results of 0.003 ng/day. The dashed line is for adipose tissue concentration and the solid line is for liver tissue concentration (ng/kg). Copyright Taylor & Francis 2014. Figure from Ruiz and co-workers, 2014 (Ruiz et al., 2014).
The immense high dose (approximately 8 mg) that was orally taken by the former president was responsible for inducing CYP1A2 metabolism (see above), causing acute liver damage. After the incident, the president was monitored for three years, measuring TCDD and its reactive metabolites in blood serum, adipose tissue, feces, skin, urine, and sweat. Because only limited human datasets are available, the available data of the president were crucial to optimize the PBPK model. The predicted outcomes after modeling such a high dose resulted in an overprediction of the adipose tissue concentrations. Predictions were in line with the observed data when the high expression of CYP1A2 enzymes were taken into account after acute exposure of TCDD.
A recent European initiative called ‘4FUN — The FUture of FUlly integrated human exposure assessment of chemicals: Ensuring the long-term viability and technology transfer of the EU-FUNded 2-FUN tools as standardized solution – http://www.4funproject.eu’ has the aim to model ecological and human exposure simultaneously. The ‘MERLING-Expo Tool’ is able to recreate complex exposure scenarios where both ecological and human targets are included. This platform allows to (i) predict bioaccumulation of PCBs in the aquatic food web (e.g., Venice lagoon and to (ii) model chemical intake and exposure in humans through a generic PBPK model (Giubilato et al., 2016; Radomyski et al., 2016).
Regulations and recommendations towards PBPK modeling: international guidelines and current state of affairs
“All models are wrong, some of them are useful” is a famous quote of Georges E.P. Box (Box and Draper, 1987). A model needs to be revised repeatedly. New insights with respect to (i) new biochemical mechanisms, (ii) physiological aspects and (iii) physicochemical characteristics of compounds are the driving force validating existing in silico models to ensure their predictive power. Knowledge on the human/animal anatomy and physiology has expanded a lot during the last decades. The specific field of mechanistic modeling increased in such a way that even formulation scientists in pharmaceutical companies blindly rely on these models to describe the in vivo performance of a drug in humans. On toxicology and risk assessment, the US Agency for Toxic Substances and Disease Registry’s (ATSDR) computational toxicology laboratory used the re-coded dioxin model to predict TCDD tissue concentrations at different exposure levels. The US Centers for Disease Control and Prevention (CDC) determines which environmental chemicals people have been exposed to and the amount of those chemicals in their bodies. Currently, over 300 environmental chemicals or their metabolites are measured in human samples (e.g., urine, blood, serum, breast milk, and meconium). CDC’s ‘National Biomonitoring Program’ provides information on human health effects, national surveillance data, and additional learning resources for each chemical and chemical group studied (https://www.cdc.gov/biomonitoring/environmental_chemicals.html). Collecting these data results in a large data set that provides a representative view of exposure to environmental chemicals and their reactive metabolites. The levels of selected chlorinated dioxin congeners were measured in blood samples and collected as part of the NHANES for the survey periods 1999–2000, 2001–2002, and 2003–2004. These concentrations are presented in the most recent National Report on Human Exposures to Environmental Chemicals (https://www.cdc.gov/biomonitoring/DioxinLikeChemicals_BiomonitoringSummary.html).
The refined model of Ruiz et al. successfully reproduced the National Health and Nutrition Examination Survey (NHANES) 2001–2002 TCDD data in age groups from 6 to 60 years and older, as well as in other human datasets (Ruiz et al., 2014). The model also enabled estimating lipid-normalized serum TCDD concentrations in breastfed infants. This validated model will be part of a toolkit which consists of a series of PBPK/PK models for volatile organic chemicals, metals and other general pollutants.
Besides the USA, Europe also demonstrates great efforts to get grip on exposure to chemical pollutants (including PCBs) using a PBPK modeling approach.
As stated by the website, the 4FUN project, funded under the EU 7th Framework Programme, aims at delivering a standardized tool for human exposure assessment to chemicals, named MERLIN-Expo. The MERLIN-Expo tool integrates on the same platform multimedia, PBPK, and dose-response models, allowing to cover all the exposure assessment chain (from concentration in water, air and/or soil to internal dose to target organs and eventually pathology risks) (Giubilato et al., 2016; Radomyski et al., 2016). In this way, it will be possible to carry out lifetime risk assessments for different human populations including exposure through multiple pathways. The long-term viability of the tool will be guaranteed analyzing the compliance of the software with relevant criteria, performing benchmarking activities, analyzing the marketing environment, formulating a business plan, and identifying stakeholder requirements. Furthermore, standardization activities and training courses ensure respectively the transparent applicability and technology transfer of the software (http://www.4funproject.eu).
Applying PBPK modeling to estimate the potential risk for human health is a more rational approach and scientifically-based framework than applying a conventional correction factor without taking into account the mechanisms of PK and PD of the compound following human exposure. PBPK modeling is a time- and cost-saving platform that will assist biologists, toxicologists and regulatory authorities in generating more rational and scientific insights in the dose-response relationships of hazardous chemicals. From this perspective, PBTK/TD modeling can serve as an in silico biomonitoring tool to secure public safety. This may assist regulatory authorities to estimate the risk towards public health after an incident with hazardous pollutants. The ‘National Biomonitoring Program’ from CDC is a step closer in towards safeguarding public health. PBPK/PBTK biomonitoring should be recognized worldwide as a standard for people’s exposure to toxic substances and for responding to serious environmental public health issues. The number of cases that use PBPK/PBTK models to estimate, define, and describe the disposition of chemicals in the human body increases. The ‘World Health Organization (WHO)’ described clearly in a 2010 document the meaning and usefulness of PBPK modeling with respect to risk assessment (World Health Organization, 2010):
- “PBPK models provide a documentable and scientifically defensible means of bridging the gap between critical toxicity studies and human risk estimates by facilitating interspecies, interindividual, high dose to low dose and route-to-route extrapolations. However, these models will not remove all of the uncertainty associated with the risk assessment process; specifically, in most cases, they would not address TD uncertainty.”
There is more trust with respect to predictions towards toxicokinetics (what the human body does with the drug) compared to toxicodynamics (what the drug does to the body). Highly likely this is because of the fact that more PK data are available (e.g., systemic/ tissue concentrations) compared to PD data which is directly related to exposure of the chemical.
- “While complex PBPK models may be relevant to chemicals for which margin between exposure and effect is small, simpler models might be adequate for preliminary assessments to inform additional steps.”
Keep it as simple as possible.
- “Comparison of model predictions with PK data is not the only way of establishing confidence in a PBPK model.”
Post-hoc sensitivity analyses are also of utmost importance to unravel the underlying
factors that may cause even higher systemic exposure for certain individuals.
- “The documentation should be sufficient to enable an experienced modeler, expert reviewer or interested end user to evaluate a PBPK model and reproduce the input–output relationships for the dose metric of relevance to the risk assessment. Transparency could be improved through development of dedicated repositories for data, models and their detailed documentation.”
Consortiums/initiatives/collaborations as described in this paragraph are indispensable to meet this WHO guideline.
- “Communication between the modeler and the risk assessor is of prime importance in developing PBPK models applicable for risk assessment.”
It should be noted that the risk assessor right from the problem formulation stage would be key in helping the modeler consider and address issues of relevance to mode of actions and risk assessment.
- “Mechanisms for adequate peer engagement at the international level for evaluating PBPK models in the context of their suitability for specific applications in risk assessment would be essential. For example, an international steering committee or a peer review group could facilitate such a process.”
Based on a joint EPAA – EURL ECVAM (European Union Reference Laboratory for Alternatives to Animal Testing) expert meeting, specialists in the field agreed on in total five strategic or more methodological high-priority recommendations, relevant to further use and acceptance of non-animal based PBTK tools (Bessems et al., 2014):
- Set up databases and collect and store human chemical-specific kinetic data.
- Establish a permanent international group of PBTK model reviewing experts.
- Develop free-to-use, readily accessible PBTK modelling web applications.
- Develop in vitro tools for high-throughput measurement of partitioning and expand the applicability domains for various tools such as in vitro absorption methods.
- Develop high-throughput and low cost analytical facilities to measure chemicals in physiological media.
Conclusion & future directions
This manuscript discusses the interesting and promising applications of PBPK modeling of hazardous PCBs to better understand their impact on human health. Modeling can help to understand how hazardous chemicals behave in the body and how one can differentiate between toxic and non-toxic effects. However, to fully rely on these models, validation and further optimization (based on new insights with respect to biochemistry and physiology) are indispensable. An interesting approach to build a reliable and predictive PBPK model is by following the guidelines presented by Sager et al. (Table 1).
Table 1: List of relevant details to report for the publication of PBPK models (Sager et al., 2015). Copyright PMC 2015.
|
Modeling workflow |
Suggested information |
|
Objectives |
What is the purpose of the model? |
|
Model acceptance criteria |
What criteria are being used to determine if a model is fit-for-purpose? |
|
What is the clinical relevance of these criteria? |
|
|
What independent data sets are used for model testing? |
|
|
Model development |
Was the model built using a PBPK software package? If not, information regarding the model structure, the source of parameters, and their physiologic context should be reported. |
|
What parameters, if any, were estimated when using parameter estimation or sensitivity analysis? |
|
|
Are the estimated parameters physiologically plausible? |
|
|
Are the parameters within the range of previously reported values (if applicable)? |
|
|
Population demographics (Do the simulated and observed populations and study sizes match?) |
|
|
Model outcomes Model performance |
Comparison of the predicted and observed PK |
|
Do the predictions meet the predetermined model specification criteria? |
|
|
Model performance |
Was sensitivity analysis performed to assess whether model output parameters are sensitive to specific input parameters? (Yes/No) |
|
What are the verified applications of the model? |
Whenever human biomonitoring data are available, these data should be used as a reference to evaluate the predictive power of the model. The more rational and scientific approach to assess the risk of hazardous chemicals will likely be adapted by regulatory authorities to demonstrate the promising value of in silico/computational modeling. In general, a better understanding of the intrinsic and extrinsic factors affecting exposure to chemicals and the knowledge of their variability and covariates, should significantly improve the ability to predict the inter-subject variability of exposure to these hazardous pollutants (Kostewicz et al., 2014). From our point of view, these mechanistic PBPK models provide the most appropriate framework within which this can be fully investigated. In that way, the value of in silico biomonitoring can be recognized as a valuable tool to safeguard public health and to respond immediately to serious environmental public health issues.
Acknowledgments
Bart Hens is aacknowledges the Flemish Research Council (FWO – Applicant number: 12R2119N).
Author Contributions
Both Luc and Bart wrote this review entitled ‘Physiologically-based Pharmacokinetic (PBPK) Modeling As a Supportive Tool to Evaluate the Impact of Polychlorinated Biphenyls (PCBs) on Public Health and Risk Assessment: Methodology, Challenges and Regulatory Perspectives’.
Conflict of interest
Authors declare no conflict of interest.
References
Agoram, B., Woltosz, W.S., Bolger, M.B., 2001. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv. Drug Deliv. Rev. 50 Suppl 1, S41-67.
Andersen, M.E., 1995. Development of physiologically based pharmacokinetic and physiologically based pharmacodynamic models for applications in toxicology and risk assessment. Toxicol. Lett. 79, 35–44.
Andersen, M.E., Clewell, H.J., Gargas, M.L., Smith, F.A., Reitz, R.H., 1987. Physiologically based pharmacokinetics and the risk assessment process for methylene chloride. Toxicol. Appl. Pharmacol. 87, 185–205.
Andersen, M.E., Mills, J.J., Gargas, M.L., Kedderis, L., Birnbaum, L.S., Neubert, D., Greenlee, W.F., 1993. Modeling receptor-mediated processes with dioxin: implications for pharmacokinetics and risk assessment. Risk Anal. 13, 25–36.
Aoki, Y., 2001. Polychlorinated biphenyls, polychlorinated dibenzo-p-dioxins, and polychlorinated dibenzofurans as endocrine disrupters--what we have learned from Yusho disease. Environ. Res. 86, 2–11. https://doi.org/10.1006/enrs.2001.4244
Aylward, L.L., Brunet, R.C., Starr, T.B., Carrier, G., Delzell, E., Cheng, H., Beall, C., 2005. Exposure reconstruction for the TCDD-exposed NIOSH cohort using a concentration- and age-dependent model of elimination. Risk Anal. 25, 945–956. https://doi.org/10.1111/j.1539-6924.2005.00645.x
Berggren, S., Gall, C., Wollnitz, N., Ekelund, M., Karlbom, U., Hoogstraate, J., Schrenk, D., Lennernäs, H., 2007. Gene and protein expression of P-glycoprotein, MRP1, MRP2, and CYP3A4 in the small and large human intestine. Mol. Pharm. 4, 252–257. https://doi.org/10.1021/mp0600687
Bessems, J.G., Loizou, G., Krishnan, K., Clewell, H.J., Bernasconi, C., Bois, F., Coecke, S., Collnot, E.-M., Diembeck, W., Farcal, L.R., Geraets, L., Gundert-Remy, U., Kramer, N., Küsters, G., Leite, S.B., Pelkonen, O.R., Schröder, K., Testai, E., Wilk-Zasadna, I., Zaldívar-Comenges, J.-M., 2014. PBTK modelling platforms and parameter estimation tools to enable animal-free risk assessment: Recommendations from a joint EPAA – EURL ECVAM ADME workshop. Regulatory Toxicology and Pharmacology 68, 119–139. https://doi.org/10.1016/j.yrtph.2013.11.008
Binnington, M.J., Curren, M.S., Quinn, C.L., Armitage, J.M., Arnot, J.A., Chan, H.M., Wania, F., 2016. Mechanistic polychlorinated biphenyl exposure modeling of mothers in the Canadian Arctic: the challenge of reliably establishing dietary composition. Environ Int 92–93, 256–268. https://doi.org/10.1016/j.envint.2016.04.011
Bischoff, K.B., Dedrick, R.L., 1968. Thiopental pharmacokinetics. J Pharm Sci 57, 1346–1351.
Bischoff, K.B., Dedrick, R.L., Zaharko, D.S., Longstreth, J.A., 1971. Methotrexate pharmacokinetics. J Pharm Sci 60, 1128–1133.
Bock, K.W., 2013. The human Ah receptor: hints from dioxin toxicities to deregulated target genes and physiological functions. Biol. Chem. 394, 729–739. https://doi.org/10.1515/hsz-2012-0340
Box, G.E.P., Draper, N.R., 1987. Empirical model-building and response surfaces, Empirical model-building and response surfaces. John Wiley & Sons, Oxford, England.
Brennan, J.C., He, G., Tsutsumi, T., Zhao, J., Wirth, E., Fulton, M.H., Denison, M.S., 2015. Development of Species-Specific Ah Receptor-Responsive Third Generation CALUX Cell Lines with Enhanced Responsiveness and Improved Detection Limits. Environ. Sci. Technol. 49, 11903–11912. https://doi.org/10.1021/acs.est.5b02906
Canaparo, R., Nordmark, A., Finnström, N., Lundgren, S., Seidegård, J., Jeppsson, B., Edwards, R.J., Boobis, A.R., Rane, A., 2007. Expression of cytochromes P450 3A and P-glycoprotein in human large intestine in paired tumour and normal samples. Basic Clin. Pharmacol. Toxicol. 100, 240–248. https://doi.org/10.1111/j.1742-7843.2006.00023.x
Carrier, G., Brunet, R.C., Brodeur, J., 1995. Modeling of the toxicokinetics of polychlorinated dibenzo-p-dioxins and dibenzofurans in mammalians, including humans. II. Kinetics of absorption and disposition of PCDDs/PCDFs. Toxicol. Appl. Pharmacol. 131, 267–276. https://doi.org/10.1006/taap.1995.1069
Chen, H.-S.G., Gross, J.F., 1979. Physiologically based pharmacokinetic models for anticancer drugs. Cancer Chemother. Pharmacol. 2, 85–94. https://doi.org/10.1007/BF00254079
Cogliano, V.J., 1998. Assessing the cancer risk from environmental PCBs. Environ Health Perspect 106, 317–323.
Cohen Hubal, E.A., Wetmore, B.A., Wambaugh, J.F., El-Masri, H., Sobus, J.R., Bahadori, T., 2019. Advancing internal exposure and physiologically-based toxicokinetic modeling for 21st-century risk assessments. J Expo Sci Environ Epidemiol 29, 11–20. https://doi.org/10.1038/s41370-018-0046-9
Edelman, I.S., Leibman, J., 1959. Anatomy of body water and electrolytes. Am. J. Med. 27, 256–277.
Englund, G., Rorsman, F., Rönnblom, A., Karlbom, U., Lazorova, L., Gråsjö, J., Kindmark, A., Artursson, P., 2006. Regional levels of drug transporters along the human intestinal tract: co-expression of ABC and SLC transporters and comparison with Caco-2 cells. Eur J Pharm Sci 29, 269–277. https://doi.org/10.1016/j.ejps.2006.04.010
Faroon, O., Ruiz, P., 2015. Polychlorinated biphenyls: New evidence from the last decade. Toxicol Ind Health. https://doi.org/10.1177/0748233715587849
Fisher, J.W., Campbell, J., Muralidhara, S., Bruckner, J.V., Ferguson, D., Mumtaz, M., Harmon, B., Hedge, J.M., Crofton, K.M., Kim, H., Almekinder, T.L., 2006. Effect of PCB 126 on hepatic metabolism of thyroxine and perturbations in the hypothalamic-pituitary-thyroid axis in the rat. Toxicol. Sci. 90, 87–95. https://doi.org/10.1093/toxsci/kfj069
Giubilato, E., Radomyski, A., Critto, A., Ciffroy, P., Brochot, C., Pizzol, L., Marcomini, A., 2016. Modelling ecological and human exposure to POPs in Venice lagoon. Part I - Application of MERLIN-Expo tool for integrated exposure assessment. Sci. Total Environ. 565, 961–976. https://doi.org/10.1016/j.scitotenv.2016.04.146
Haggard, H.W., 1924. The Absorption, Distribution, and Elimination of Ethyl Ether Ii. Analysis of the Mechanism of Absorption and Elimination of Such a Gas or Vapor as Ethyl Ether. J. Biol. Chem. 59, 753–770.
Hansen, L.G., 2012. The ortho Side of PCBs: Occurrence and Disposition. Springer Science & Business Media.
Hens, B., Dyke, P., Hens, L., 2016. What can we learn from ‘dioxin incidents’? IJEP 60, 34–62. http://dx.doi.org/10.1504/IJEP.2016.10002954
Hens, B., Hens, L., 2017. Persistent Threats by Persistent Pollutants: Chemical Nature, Concerns and Future Policy Regarding PCBs-What Are We Heading For? Toxics 6. https://doi.org/10.3390/toxics6010001
Holland, E.B., Feng, W., Zheng, J., Dong, Y., Li, X., Lehmler, H.-J., Pessah, I.N., 2017. An Extended Structure–Activity Relationship of Nondioxin-Like PCBs Evaluates and Supports Modeling Predictions and Identifies Picomolar Potency of PCB 202 Towards Ryanodine Receptors. Toxicol Sci 155, 170–181. https://doi.org/10.1093/toxsci/kfw189
Jamei, M., Marciniak, S., Feng, K., Barnett, A., Tucker, G., Rostami-Hodjegan, A., 2009. The Simcyp population-based ADME simulator. Expert Opin Drug Metab Toxicol 5, 211–223. https://doi.org/10.1517/17425250802691074
Kedderis, L.B., Mills, J.J., Andersen, M.E., Birnbaum, L.S., 1993. A physiologically based pharmacokinetic model for 2,3,7,8-tetrabromodibenzo-p-dioxin (TBDD) in the rat: tissue distribution and CYP1A induction. Toxicol. Appl. Pharmacol. 121, 87–98. https://doi.org/10.1006/taap.1993.1132
Kohn, M.C., Lucier, G.W., Clark, G.C., Sewall, C., Tritscher, A.M., Portier, C.J., 1993. A mechanistic model of effects of dioxin on gene expression in the rat liver. Toxicol. Appl. Pharmacol. 120, 138–154. https://doi.org/10.1006/taap.1993.1096
Kostewicz, E.S., Aarons, L., Bergstrand, M., Bolger, M.B., Galetin, A., Hatley, O., Jamei, M., Lloyd, R., Pepin, X., Rostami-Hodjegan, A., Sjögren, E., Tannergren, C., Turner, D.B., Wagner, C., Weitschies, W., Dressman, J., 2014. PBPK models for the prediction of in vivo performance of oral dosage forms. Eur J Pharm Sci 57, 300–321. https://doi.org/10.1016/j.ejps.2013.09.008
Larsen, J.C., 2006. Risk assessments of polychlorinated dibenzo- p-dioxins, polychlorinated dibenzofurans, and dioxin-like polychlorinated biphenyls in food. Mol Nutr Food Res 50, 885–896. https://doi.org/10.1002/mnfr.200500247
Lee, S.K., Ou, Y.C., Yang, R.S.H., 2002. Comparison of pharmacokinetic interactions and physiologically based pharmacokinetic modeling of PCB 153 and PCB 126 in nonpregnant mice, lactating mice, and suckling pups. Toxicol. Sci. 65, 26–34.
Li, Y.F., Zhang, C., Zhou, S., He, M., Zhang, H., Chen, N., Li, F., Luan, X., Pai, M., Yuan, H., Sun, D., Li, Y., 2018. Species difference in paclitaxel disposition correlated with poor pharmacological efficacy translation from mice to humans. Clin Pharmacol 10, 165–174. https://doi.org/10.2147/CPAA.S185449
Lind, P.M., Orberg, J., Edlund, U.-B., Sjöblom, L., Lind, L., 2004. The dioxin-like pollutant PCB 126 (3,3’,4,4’,5-pentachlorobiphenyl) affects risk factors for cardiovascular disease in female rats. Toxicol. Lett. 150, 293–299. https://doi.org/10.1016/j.toxlet.2004.02.008
Loftsson, T., 2015. Essential Pharmacokinetics: A Primer for Pharmaceutical Scientists. Academic Press.
Lukacova, V., Woltosz, W.S., Bolger, M.B., 2009. Prediction of modified release pharmacokinetics and pharmacodynamics from in vitro, immediate release, and intravenous data. AAPS J 11, 323–334. https://doi.org/10.1208/s12248-009-9107-2
Masuda, Y., 1996. Approach to risk assessment of chlorinated dioxins from Yusho PCB poisoning. Chemosphere 32, 583–594.
Mills, J.J., Andersen, M.E., 1993. Dioxin hepatic carcinogenesis: biologically motivated modeling and risk assessment. Toxicol. Lett. 68, 177–189.
Moffett, D.B., El-masri, H.A., Fowler, BRUCE A., 2007. CHAPTER 6 - General Considerations of Dose-Effect and Dose-Response Relationships**This chapter is based on the chapter with the same title by Emil A. Pfitzer and Velimir Vouk in the 2nd edition of the Handbook.Disclaimer: The findings and conclusions in this chapter are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention/Agency for Toxic Substances and Disease Registry or the Environmental Protection Agency., in: Nordberg, G.F., Fowler, Bruce A., Nordberg, M., Friberg, L.T. (Eds.), Handbook on the Toxicology of Metals (Third Edition). Academic Press, Burlington, pp. 101–115. https://doi.org/10.1016/B978-012369413-3/50061-6
Morin, R.L., 2017. Monte Carlo Simulation: A Ubiquitous Tool. J Am Coll Radiol 14, 416–417. https://doi.org/10.1016/j.jacr.2016.12.014
National Research Council, 2001. Drinking Water and Health, Volume 8: Pharmacokinetics in Risk Assessment. https://doi.org/10.17226/1015
Ozkaynak, H., Frey, H.C., Hubbell, B., 2008. Characterizing Variability and Uncertainty in Exposure Assessments Improves Links to Environmental Decision-Making. EM (Pittsburgh Pa) 58, 18–22.
Parham, F.M., Kohn, M.C., Matthews, H.B., DeRosa, C., Portier, C.J., 1997. Using structural information to create physiologically based pharmacokinetic models for all polychlorinated biphenyls. Toxicol. Appl. Pharmacol. 144, 340–347.
Pearce, R., Setzer, R.W., Davis, J., Wambaugh, J.F., 2017. Evaluation and calibration of high-throughput predictions of chemical distribution to tissues. J Pharmacokinet Pharmacodyn 44, 549–565. https://doi.org/10.1007/s10928-017-9548-7
Peyret, T., Poulin, P., Krishnan, K., 2010. A unified algorithm for predicting partition coefficients for PBPK modeling of drugs and environmental chemicals. Toxicol. Appl. Pharmacol. 249, 197–207. https://doi.org/10.1016/j.taap.2010.09.010
Pizzo, F., Benfenati, E., 2016. In Silico Models for Repeated-Dose Toxicity (RDT): Prediction of the No Observed Adverse Effect Level (NOAEL) and Lowest Observed Adverse Effect Level (LOAEL) for Drugs. Methods Mol. Biol. 1425, 163–176. https://doi.org/10.1007/978-1-4939-3609-0_9
Radomyski, A., Giubilato, E., Ciffroy, P., Critto, A., Brochot, C., Marcomini, A., 2016. Modelling ecological and human exposure to POPs in Venice lagoon - Part II: Quantitative uncertainty and sensitivity analysis in coupled exposure models. Sci. Total Environ. 569–570, 1635–1649. https://doi.org/10.1016/j.scitotenv.2016.07.057
Rescigno, A., 1997. FUNDAMENTAL CONCEPTS IN PHARMACOKINETICS. Pharmacological Research 35, 363–390. https://doi.org/10.1006/phrs.1997.0175
Rotroff, D.M., Wetmore, B.A., Dix, D.J., Ferguson, S.S., Clewell, H.J., Houck, K.A., Lecluyse, E.L., Andersen, M.E., Judson, R.S., Smith, C.M., Sochaski, M.A., Kavlock, R.J., Boellmann, F., Martin, M.T., Reif, D.M., Wambaugh, J.F., Thomas, R.S., 2010. Incorporating human dosimetry and exposure into high-throughput in vitro toxicity screening. Toxicol. Sci. 117, 348–358. https://doi.org/10.1093/toxsci/kfq220
Rowland, M., Peck, C., Tucker, G., 2011a. Physiologically-Based Pharmacokinetics in Drug Development and Regulatory Science. Annu. Rev. Pharmacol. Toxicol. 51, 45–73. https://doi.org/10.1146/annurev-pharmtox-010510-100540
Rowland, M., Tozer, T.N., Rowland, M., 2011b. Clinical pharmacokinetics and pharmacodynamics: concepts and applications, 4th ed. ed. Wolters Kluwer Health/Lippincott William & Wilkins, Philadelphia.
Ruiz, P., Aylward, L.L., Mumtaz, M., 2014. Application of pharmacokinetic modelling for 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure assessment. SAR QSAR Environ Res 25, 873–890. https://doi.org/10.1080/1062936X.2014.962083
Sager, J.E., Yu, J., Ragueneau-Majlessi, I., Isoherranen, N., 2015. Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation Approaches: A Systematic Review of Published Models, Applications, and Model Verification. Drug Metab Dispos 43, 1823–1837. https://doi.org/10.1124/dmd.115.065920
Sasso, A.F., Georgopoulos, P.G., Isukapalli, S.S., Krishnan, K., 2012. Bayesian Analysis of a Lipid-Based Physiologically Based Toxicokinetic Model for a Mixture of PCBs in Rats. J Toxicol 2012. https://doi.org/10.1155/2012/895391
Schmitt, W., 2008. General approach for the calculation of tissue to plasma partition coefficients. Toxicol In Vitro 22, 457–467. https://doi.org/10.1016/j.tiv.2007.09.010
Sharma, R.P., Schuhmacher, M., Kumar, V., 2018. The development of a pregnancy PBPK Model for Bisphenol A and its evaluation with the available biomonitoring data. Sci. Total Environ. 624, 55–68. https://doi.org/10.1016/j.scitotenv.2017.12.023
Sipes, N.S., Wambaugh, J.F., Pearce, R., Auerbach, S.S., Wetmore, B.A., Hsieh, J.-H., Shapiro, A.J., Svoboda, D., DeVito, M.J., Ferguson, S.S., 2017. An Intuitive Approach for Predicting Potential Human Health Risk with the Tox21 10k Library. Environ. Sci. Technol. 51, 10786–10796. https://doi.org/10.1021/acs.est.7b00650
Sorg, O., Zennegg, M., Schmid, P., Fedosyuk, R., Valikhnovskyi, R., Gaide, O., Kniazevych, V., Saurat, J.-H., 2009. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) poisoning in Victor Yushchenko: identification and measurement of TCDD metabolites. Lancet 374, 1179–1185. https://doi.org/10.1016/S0140-6736(09)60912-0
Teorell, T., 1937a. Kinetics of distribution of substances administered to the body, I : The extravascular modes of administration. Archives internationales de pharmacodynamie et de therapie 57, 205–225.
Teorell, T., 1937b. Kinetics of distribution of substances administered to the body, II : The intravascular modes of administration. Archives internationales de pharmacodynamie et de therapie 57, 226–240.
Ulaszewska, M.M., Ciffroy, P., Tahraoui, F., Zeman, F.A., Capri, E., Brochot, C., 2012. Interpreting PCB levels in breast milk using a physiologically based pharmacokinetic model to reconstruct the dynamic exposure of Italian women. J Expo Sci Environ Epidemiol 22, 601–609. https://doi.org/10.1038/jes.2012.36
Van den Berg, M., Birnbaum, L., Bosveld, A.T., Brunström, B., Cook, P., Feeley, M., Giesy, J.P., Hanberg, A., Hasegawa, R., Kennedy, S.W., Kubiak, T., Larsen, J.C., van Leeuwen, F.X., Liem, A.K., Nolt, C., Peterson, R.E., Poellinger, L., Safe, S., Schrenk, D., Tillitt, D., Tysklind, M., Younes, M., Waern, F., Zacharewski, T., 1998. Toxic equivalency factors (TEFs) for PCBs, PCDDs, PCDFs for humans and wildlife. Environ. Health Perspect. 106, 775–792.
Wagner, J.G., 1981. History of pharmacokinetics. Pharmacol. Ther. 12, 537–562.
Weijs, L., Roach, A.C., Yang, R.S.H., McDougall, R., Lyons, M., Housand, C., Tibax, D., Manning, T., Chapman, J., Edge, K., Covaci, A., Blust, R., 2014. Lifetime PCB 153 bioaccumulation and pharmacokinetics in pilot whales: Bayesian population PBPK modeling and Markov chain Monte Carlo simulations. Chemosphere 94, 91–96. https://doi.org/10.1016/j.chemosphere.2013.09.019
Willmann, S., Thelen, K., Lippert, J., 2012. Integration of dissolution into physiologically-based pharmacokinetic models III: PK-Sim®. J. Pharm. Pharmacol. 64, 997–1007. https://doi.org/10.1111/j.2042-7158.2012.01534.x
Wood, S.A., Armitage, J.M., Binnington, M.J., Wania, F., 2016. Deterministic modeling of the exposure of individual participants in the National Health and Nutrition Examination Survey (NHANES) to polychlorinated biphenyls. Environ Sci Process Impacts 18, 1157–1168. https://doi.org/10.1039/c6em00424e
World Health Organization (Ed.), 2010. Characterization and application of physiologically based pharmacokinetic models in risk assessment, Harmonization project document. World Health Organization, Geneva.
Zhao, P., Zhang, L., Grillo, J.A., Liu, Q., Bullock, J.M., Moon, Y.J., Song, P., Brar, S.S., Madabushi, R., Wu, T.C., Booth, B.P., Rahman, N.A., Reynolds, K.S., Gil Berglund, E., Lesko, L.J., Huang, S.-M., 2011. Applications of Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation During Regulatory Review. Clinical Pharmacology & Therapeutics 89, 259–267. https://doi.org/10.1038/clpt.2010.298
Zhu, X.-W., Sedykh, A., Zhu, H., Liu, S.-S., Tropsha, A., 2013. The use of pseudo-equilibrium constant affords improved QSAR models of human plasma protein binding. Pharm. Res. 30, 1790–1798. https://doi.org/10.1007/s11095-013-1023-6

