Rozlana idiopatyczna hiperostoza szkieletu (DISH) jest stanem charakteryzującym się zwapnieniem i kostnieniem więzadeł kręgosłupa szyjnego; w niektórych przypadkach może to skutkować dysfagią.
- cervical spine
- DISH
- diffuse idiopathic skeletal hyperostosis
- Forestier disease
- dysphagia
- molecular and genetical factors
- DISHphagia
1. Wstęp
Diffuse idiopathic skeletal hyperostosis (DISH/Forestier’s disease) is a condition characterized by the calcification and ossification of the ligaments of the cervical spine, and the condition may be exclusive to this area of the spine [1].
DISH occurs with a frequency of 2–4% in patients over 40 years of age, up to 11% with middle-aged patients, and this increases to 28% in those over 80 years of age [2]. DISH is more common in men [2,3][2][3].
DISH is often confused with related major diseases such as ankylosing spondylitis, spondylosis deformans, and degenerative spinal disease [3].
2. Etiopathogenesis—Molecular Aspects
Little is known about the pathogenesis of DISH. The condition is associated with old age, males, obesity, hypertension, atherosclerosis, and diabetes mellitus [4]. On the molecular level, the condition is associated with genetic factors, changes in signaling pathways, and metabolic and vascular [5] inflammatory [6] factors.
The pathogenesis of DISH is likely polygenic and dependent on the interaction of multiple gene variants, epigenetics, and environmental factors. DISH is likely influenced by many polymorphisms influencing inheritance, pathology, and expression in various genes [7].
2.1. Genetic Factors
Many proteins and signaling pathways are responsible for bone formation and healing, including the Wnt/β-catenin signaling pathway. Wnt ligands bind to the receptor complex (frizzled and low-density lipoprotein receptor-related protein (LRP5 or LPR6)), causing the release of β-catenin by phosphorylation, which releases it from the multiprotein complex. The accumulation of β-catenin in the cytoplasm and ultimately its translocation to the nucleus result in the activation of target genes [8]. TGF-β is another signaling pathway involved in the differentiation of bone-forming osteoblasts, skeletal genesis, and bone homeostasis. In humans, heterozygous pathogenic changes in fibrillin 1 (FBN1), transforming growth factor beta receptor 1/2 (TGFBR1/2), or in TGF-β inhibit this signaling pathway, phenotypically causing skeletal-deformity diseases (Marfan, Loeys Dietz, and Camurati–Engelman syndrome) [9]. Many cytokines, growth factors, hormones, and vitamins are involved in the bone-repair and -remodeling phases. Growth factors belonging to the TGF-β superfamily, bone morphogenetic proteins (BMP), or TGF-β1 act locally on bone formation by stimulating the proliferation and osteogenic differentiation of mesenchymal stem cells (MSC) [9].
Gorman et al. described a family with a genetic predisposition to DISH of the cervical spine [10]. Among animals, some dog breeds (for example, boxers) were found to have a higher incidence of DISH (40%) than that of other breeds (4%) [11]. Previous research found that mouse modeling lacked the equivalent volume of nucleoside transporter 1 (ENT1) signaling DISH [12].
Single nucleotide polymorphisms in the COL6A1 and FGF2 genes were associated with the presence of DISH [12]. Other potential genes that were studied in patients with DISH and chondrocalcinosis (DISH/CC) include R-spondin 4 (RSPO4), LEM domain-containing 3 (LEMD3), and protein phosphatase 2 regulatory subunit beta delta 2 (PPPR2RD) (Table 1, Figure 1) [13,14][13][14].
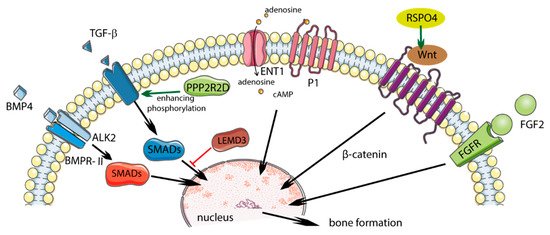
Figure 1. Potential role of polymorphism in diffuse idiopathic skeletal hyperostosis (DISH) and influence on regulatory pathways. Solid green arrows indicate activation; solid red bars indicate inhibition. Figure created using Servier Medical Art: https://smart.servier.com (accessed on 4 February 2021) [17][15].
Table 1. Genetic variants associated with DISH.
| Gene | SNP | Population | Disorder Associated with DISH | References |
|---|---|---|---|---|
| FGF2 | rs1476217/rs3747676 | Korean | OPLL | Jun et al. [15][16] |
| COL6A1 | intron 32 (−29) | Japanese | OPLL | Tsukahara et al. [16][17] |
| PPP2R2D | rs34473884 | Azorean | CC | Parreira et al. [14] |
| RSPO4 | rs146447064, rs14915407 | Azorean | CC | Couto er al. [13] |
| BMP4 | rs17563 | Azorean | CC | Couto er al. [13] |
| LEMD3 | rs201930700 | Azorean | CC | Couto er al. [13] |
SNP, single nucleotide polymorphism; OPLL, ossification of the posterior longitudinal ligament; CC, chondrocalcinosis. Future molecular studies should include other variants of the BMP4 gene (rs17563) and the overexpression of ABCC6 [7].
Future molecular studies should include other variants of the BMP4 gene (rs17563) and the overexpression of ABCC6 [7].
2.1.1. COL6A1
The α-chain of Type VI collagen is a protein of the extracellular matrix encoded by the COL6A1 gene, which forms the basis of osteoblastic cells or chondrocytes, which participate in membrane or endochondral ossification. Certain COL6A1 variants were shown to increase the posterior longitudinal ligament’s (OPLL) susceptibility to ossification. Single-nucleotide polymorphisms (SNPs) in COL6A1 were only found in the Japanese DISH patient population [16][17]. Patients with both OPLL and DISH showed more potent COL6A1 variants than patients with isolated OPLL did [12].
2.1.2. FGF2
Fibroblast growth factor 2 (FGF2) is part of a large family of growth factors that play a role in the angiogenesis and mitogenesis of many cell types. FGF2 transmits signals through two (FGFR2 and FGFR3) of the four known FGF receptors (FGFR), which contain an extracellular immunoglobulin-like domain and a cytoplasmic tyrosine kinase domain. The FGF family of proteins are involved in stem-cell growth, tissue regeneration, and carcinogenesis [18]. The signaling pathways of FGF/FGFR play an important role in bone development. In addition, FGF signaling is involved in the maintenance of adult bone homeostasis and fracture healing. The synergism of FGF2 action with BMP2 with canonical Wnt signaling was demonstrated [19].
FGF/FGFR plays a major role in bone development, including in the pathogenesis of human ligament ossification. Previous research showed that the variant of FGFR1 is significantly associated with OPLL, and another variant in FGF2 is associated with conditions coexisting with DISH [16][17].
2.1.3. RSPO4
Proteins from the R-spondin (RSPO) family consist of four members (RSpo1–4) whose structures are 60% similar. The RSPO4 protein is involved in the activation of the Wnt/β-catenin signaling pathways. The activation of the Wnt signaling pathway causes the production of proteins responsible for bone formation, while disabling this pathway reduces the production of bone tissue. In patients with DISH/CC, two variants of the RSPO4 gene (rs146447064 and rs14915407) were found, and they occurred more often than in the control group. Reducing the expression of the RSPO4 gene decreases Wnt activation and may “protect” against the supergrowth of new bone [14].
2.1.4. LEMD3
The LEMD3 gene encodes 60 kD inner nuclear membrane protein Man1 (LEMD3) and is a nuclear membrane protein that can inhibit the TGFβ signaling pathway by binding to SMAD proteins, which are downstream pathway mediators. The heterozygous loss of function mutations in LEMD3 enhances TGF-β signaling and leads to bone developmental disorders such as osteopoikilosis and melorheostosis. LEMD3 normally antagonizes the activation of the bone lining to bone-forming osteoblasts, generating a modeling-based formation, possibly by reducing BMP and/or TGF-β signaling. Pathogenic LEMD3 variants can disrupt the antagonistic effect of LEMD3 on TGF-β and BMP signaling, resulting in the increased proliferation and early differentiation of bone-lining cells [20].
A previously discovered rare LEMD3 variant (rs201930700) of the DISH/CC phenotype may promote enhanced TGF-β signaling, leading to increased bone formation [14].
2.1.5. PPP2R2D
Protein phosphatases influence the signaling of the TGF-β superfamily, which regulates numerous cellular responses. They catalyze the removal of phosphate groups from serine and/or threonine residues by the hydrolysis of phosphoric acid monoesters. PPP2R2D protein inhibits Type I ALK4 and ALK5 receptors in human cell lines, causing the blockage of TGF-β pathway signaling. Parreira et al. found that the PPP2R2D variant (rs34473884), which was significantly correlated with the DISH/CC phenotype, is the likely cause of the condition’s development [15][16].
2.1.6. BMP4
BMP4 is a member of the BMP family and the TGF-β superfamily. It is involved in osteoblast differentiation and bone formation, and is a factor that stimulates soft bone ossification and the healing of bone fractures at an early stage. BMP4 was also found in mesenchymal cells, soft bone cells, the periosteum, bone-marrow cavities, and muscle cells adjacent to a fractured bone. BMP4 can indirectly stimulate bone formation by inhibiting osteoclastogenesis.
The increased expression of BMP2 and BMP4 mRNA was observed as a result of mechanical stress in spinal ligament cells in OPLL patients, and it is therefore possible that this pathway may also be involved in ossification in DISH [5]. A significant association was identified between DISH/CC and the genetic variant in BMP4 (rs17563) [7].
2.1.7. ALK2
Activin type I receptor (ALK2) is a transmembrane serine kinase receptor that activates intracellular signaling pathways when bound to BMP. The p.K400E mutation in the ALK2 gene was demonstrated in a patient with DISH. Overexpressing cells ALK2 p.K400E activate BMP signaling in response to osteogenic BMP ligands and a lack of response to non-osteogenic ligand - activin A. BMP signaling ALK2 p.K400E was further enhanced by the coexpression of the BMP type II receptor (BMPR-II) that increased phosphorylation of ALK2 p. K400E. Increased ossification in DISH may be due to the enhanced induction of osteogenic BMP signaling (more than activin A) by the ALK2 p.K400E receptors enhanced by BMPR-II [21,22][21][22].
2.1.8. ENT1
Type 1 equilibrative nucleoside transporter (ENT1) is responsible for most of the transport of adenosine across the cell membrane, which plays a role in bone homeostasis [23]. Adenosine promotes bone formation and resorption by activating different molecular pathways depending on which adenosine receptor is activated [24].
Mice lacking ENT1 (ENT1-KO) exhibited decreased adenosine uptake, increased plasma levels of circulating adenosine, and progressive ectopic calcified tissue of the spine and the annulus fibrosus of the intervertebral disc [20]. Studies were carried out on age-matched wild-type (WT) and ENT1-KO mice, and human cadaver spines meeting radiographic DISH criteria. The role of ENT1 in bone metabolism in vivo may be confirmed by the fact that patients without ENT1 exhibit increased ectopic mineralization [25].
Micro-computed tomography (CT) ENT1-KO mice revealed ectopic tissue calcification in the spine cervical–thoracic section. Histological examination of human DISH changes in mice resembled ENT1-KO. Microarray analysis in the affected tissue of ENT1-KO mice revealed extensive transcription dysregulation of genes related to the cell cycle and genes involved in the regulation of bone mineralization and development [26].
This phenomenon may explain how high levels of adenosine reduce the sensitivity of adenosine receptors involved in the regulation of osteoclast and osteoblast functions [27].
2.1.9. Epigenetics in DISH
Epigenetic changes in the Wnt pathway genes are associated with changes in bone density and osteoblast function [28]. DNA methylation is an important gene regulator of the Wnt pathway. The Wnt ROR2 coreceptor shows progressive reduction in the methylation of its promoter DNA as it matures from mesenchymal stem cells into differentiated osteoblasts. The hypomethylation of ROR2 and WNT5a was demonstrated in the mesenchymal stem cells of isolated spinal ligaments in patients with DISH. This hypomethylation was associated with increased expression of ROR2 and WNT5a [29].
Noncoding RNA (ncRNA) includes microRNA (miRNA), long noncoding RNA (lncRNA), and circular RNA (circRNA), which regulate neoplastic, inflammatory, and degenerative processes [30]. miRNAs are short noncoding RNAs that are about 22 nucleotides in length and bind to complementary targets in the three untranslated regions (UTRs) of messenger RNA (mRNA), inhibiting mRNA translation and regulating many signaling pathways [31]. The level of miRNA expression varies by tissue and fine-tunes gene expression [30]. miRNAs interact with circRNA and lncRNA, regulating their stability; in turn, they bind to miRNAs, alleviating the inhibition of miRNAs on their target genes and increasing the expression levels of target genes [31]. The participation of noncoding RNA in ossification of spinal ligament was demonstrated. miR-563 interacts with SMURF1, which contributes to the ossification of OPLL by impeding ubiquitin-independent Smad degradation [32]. The role of miR-615-3p and miR-132-3p is to inhibit osteogenic differentiation by targeting forkhead box O1 protein (FOXO1) and growth differentiation factor 5 (GDF5) [32]. Angio miR-494 and miR-126-5p contribute to the proangiogenic effects of BMP4 on endothelial cells [33]. However, ncRNA coregulation between OPLL and DISH was not described.
2.2. Metabolic Factors
Growth hormone and insulin-like growth factor 1 were shown to promote bone formation and found to be elevated in patients with DISH [5]. The proliferation of cells forming fibroblasts, myoblasts, and osteoblasts is stimulated by TGF-β1, insulin, and bone morphogenic protein (BMP2). In DISH, the mechanisms of new bone growth, especially in tendon-attachment areas, are not known, and the possible signaling pathways involved in this process are Wnt-β catenins, nuclear factor κB, BMP2, prostaglandin I2, and endothelin1 [34].
Obesity correlated with DISH may be a factor in the pathogenesis of spinal calcification through a chronic process and inflammatory mediators (IL-6, TNF alpha, TGF-beta, VEGF) due to the hypertrophy and ossification of spinal ligaments. Obesity may also significantly affect the peripheral activity of osteoblasts and osteocytes through centrally regulated processes. Leptin (adipokine) acts as a cytokine and hormone in bone metabolism and cartilage homeostasis. As a proinflammatory cytokine, it stimulates the reorganization of the chondrocyte cytoskeleton by increasing the secretion of degradation mediators, and takes part in intervertebral-disc (IVD) tissue homeostasis by stimulating the proliferation of intervertebral disc cells and inhibiting the apoptosis of nucleus cells. In degenerative intervertebral disc disease (DDD), an increase in leptin was shown in IVD and the ligamentum flavum. Leptin mediating the inflammatory response of IL-6 causes hypertrophy and fibrosis of the yellow ligament by increasing collagen production. Leptin is expressed in the yellow ligament and adjacent fat-favoring epidural fibrosis yellow ligament. Adiponectin is an adipokine with an anti-inflammatory effect that reduces the production of TNF alpha, and it is less concentrated in DDD. In addition, the control of fat in bone regulation showed caloric state-dependent responses in central neuropeptide Y (NPY). In states where high-calorie NPY decreases rapidly, it increases the activity of osteoblasts [35].
Sohn et al. demonstrated an increase in bone mineral density (BMD) of the lumbar spine and the femoral neck in patients with DISH compared to the control group. The number of spine levels covered by DISH correlated with the BMD of the lumbar spine. These results may suggest that, in DISH, the increase in BMD is a consequence of metabolic processes [36].
2.2.1. Metabolic Syndrome and Obesity
Mader et al. found statistically significantly increased waist circumference and BMI in patients with DISH compared to those of the control group, regardless of gender, which indicates an association with obesity [37]. There were no significant differences between groups in serum total cholesterol, high-density lipoprotein, low-density lipoprotein, or serum triglycerides [37,38][37][38].
2.2.2. Diabetes
There is increased incidence of DISH in people with Type 2 diabetes and impaired glucose tolerance [39,40][39][40]. The Wnt signal pathway is regulated by inhibitors sclerostin and Dickkopf-related protein-1 (Dkk-1). Patients with DISH showed lower levels of DKK-1 than those of healthy people [39].
2.2.3. Gout
Although DISH is more often diagnosed in patients with gout, genotypic analysis related to gout did not confirm that it was a cause of DISH. Most likely, gout and DISH share common risk factors, such as metabolic syndrome and obesity [41].
2.3. Vascular Factors
A significant increase in the number and width of the vertebral body nutritional orifices and hypervascularization was observed in people with DISH [42]. However, it is not known whether the newly formed bone required more supplying blood vessels or the growth of the blood vessels facilitated or caused new bone to form.
The process of heterotopic new bone formation in the tendon attachment areas of DISH patients is associated with abnormal osteoblast growth and activity, with active bone remodeling. Osteogenesis is initiated by the growth factors insulin, insulin-like growth factor-1, and growth hormone. Their action is not limited to bone tissue alone, and it is not clear why ossification only begins in certain places. One possible explanation for this selective localization is angiogenesis, which is essential for osteoblast proliferation. This may be due to the complex pathway of intercellular signaling between the vascular endothelium and bone cells involving numerous mediators such as VEGF, the primary fibroblast growth factor, TGF-β, and PDGF. This vascular endothelial mediation mechanism enables the targeting of osteoclast and osteoblast precursors to specific locations. These processes are also regulated by mediators and bone regulators including cytokines, estrogen, and parathyroid hormone (PTH). Angiogenesis is an important factor in metabolic syndrome, visceral obesity, dyslipidemia, diabetes, and atherosclerosis; the association of metabolic disorders with DISH may support the role of angiogenesis. In patients with DISH, a higher incidence of aortic-valve sclerosis (a marker of atherosclerosis and cardiovascular events) and bonelike arterial calcification was found. Due to the angiogenesis stimulating activity in patients with DISH, the disease can be considered part of the syndrome, not the disease. Metabolic disorders accompanied by DISH, and their common characteristic is angiogenesis [43 ][43].
Inny aspekt naczyniowy zbadali Bakker i wsp., którzy wykazali, że nowo utworzona kość w DISH kręgosłupa szyjnego jest zlokalizowana symetrycznie przed trzonem kręgu, w przeciwieństwie do kręgosłupa piersiowego, gdzie znajduje się asymetrycznie na przednio-bocznej powierzchni kręgosłupa. kręg. Tętnice mogą działać jako naturalna bariera dla nowo powstałej kości w DISH, więc główne naczynia w szyi znajdujące się po bokach trzonów kręgów oraz brak tętnic segmentowych w kręgach szyjnych mogą przyczyniać się do liniowego (pręcikowatego) wzrostu kości, w przeciwieństwie do klatki piersiowej. kręgosłup, w którym obecne są naczynia segmentarne. Nieograniczone tworzenie kości do przodu może wyjaśniać brzuszne przemieszczenie tchawicy i przełyku [ 44 ] [44].
2.4. Czynniki zapalne
Niektórzy autorzy sugerują, że czynniki zapalne charakteryzują DISH, ale jak dotąd nie znaleziono dowodów naukowych [ 6 ][6]. Badania histologiczne ludzkich pacjentów z DISH w porównaniu z myszami ENT1-KO nie wykazały charakteru zapalnego [ 26 ][26].
(Referencje zostaną dodane automatycznie, gdy wpis będzie dostępny online)
References
- Pappone, N.; Ambrosino, P.; di Minno, M.N.D.; Iervolino, S. Is diffuse idiopathic skeletal hyperostosis a disease or a syndrome? Rheumatology 2017, 56, 1635–1636.
- Bakker, J.T.; Kuperus, J.S.; Kuijf, H.J.; Oner, F.C.; de Jong, P.A.; Verlaan, J.J. Morphological characteristics of diffuse idiopathic skeletal hyperostosis in the cervical spine. PLoS ONE 2017, 12, e0188414.
- Mader, R.; Pappone, N.; Baraliakos, X.; Eshed, I.; Sarzi-Puttini, P.; Atzeni, F.; Bieber, A.; Novofastovski, I.; Kiefer, D.; Verlaan, J.J.; et al. Diffuse Idiopathic Skeletal Hyperostosis (DISH) and a Possible Inflammatory Component. Curr. Rheumatol. Rep. 2021, 23, 6.
- Dixon, J.; Beaucage, K.; Nagao, M.; Lajoie, G.; Veras, M.; Fournier, D.; Holdsworth, D.; Bailey, C.; Hammond, J.; Séguin, C. Mice lacking the nucleoside transporter ENT1: A model of diffuse idiopathic skeletal hyperostosis in humans. In Proceedings of the Orthopaedic Proceedings, Montreal, QC, Canada, 17 July 2020; p. 123.
- Mader, R.; Verlaan, J.J.; Buskila, D. Diffuse idiopathic skeletal hyperostosis: Clinical features and pathogenic mechanisms. Nat. Rev. Rheumatol. 2013, 9, 741–750.
- Mader, R.; Pappone, N.; Baraliakos, X.; Eshed, I.; Sarzi-Puttini, P.; Atzeni, F.; Bieber, A.; Novofastovski, I.; Kiefer, D.; Verlaan, J.J.; et al. Diffuse Idiopathic Skeletal Hyperostosis (DISH) and a Possible Inflammatory Component. Curr. Rheumatol. Rep. 2021, 23, 6.
- Parreira, B. Genetic Variants Associated with Ectopic Calcifications. Ph.D. Thesis, University of Algarve, Faro, Portugal, 2018.
- Gregson, C.L.; Duncan, E.L. The Genetic Architecture of High Bone Mass. Front. Endocrinol. 2020, 11, 595653.
- Frost, M.; Rahbek, E.T.; Ejersted, C.; Hoilund-Carlsen, P.F.; Bygum, A.; Thomsen, J.S.; Andreasen, C.M.; Andersen, T.L.; Frederiksen, A.L. Modeling-based bone formation transforms trabeculae to cortical bone in the sclerotic areas in Buschke-Ollendorff syndrome. A case study of two females with LEMD3 variants. Bone 2020, 135, 115313.
- Gorman, C.; Jawad, A.S.; Chikanza, I. A family with diffuse idiopathic skeletal hyperostosis. Ann. Rheum. Dis. 2005, 64, 1794–1795.
- Kranenburg, H.C.; Westerveld, L.A.; Verlaan, J.J.; Oner, F.C.; Dhert, W.J.; Voorhout, G.; Hazewinkel, H.A.; Meij, B.P. The dog as an animal model for DISH? Eur. Spine J. 2010, 19, 1325–1329.
- Warraich, S.; Bone, D.B.; Quinonez, D.; Ii, H.; Choi, D.S.; Holdsworth, D.W.; Drangova, M.; Dixon, S.J.; Seguin, C.A.; Hammond, J.R. Loss of equilibrative nucleoside transporter 1 in mice leads to progressive ectopic mineralization of spinal tissues resembling diffuse idiopathic skeletal hyperostosis in humans. J. Bone Miner. Res. 2013, 28, 1135–1149.
- Couto, A.R.; Parreira, B.; Thomson, R.; Soares, M.; Power, D.M.; Stankovich, J.; Armas, J.B.; Brown, M.A. Combined approach for finding susceptibility genes in DISH/chondrocalcinosis families: Whole-genome-wide linkage and IBS/IBD studies. Hum. Genome Var. 2017, 4, 17041.
- Parreira, B.; Couto, A.R.; Rocha, F.; Sousa, M.; Faustino, V.; Power, D.M.; Bruges-Armas, J. Whole exome sequencing of patients with diffuse idiopathic skeletal hyperostosis and calcium pyrophosphate crystal chondrocalcinosis. Acta Reumatol. Port. 2020, 45, 116–126.
- Servier Medical Art. Available online: (accessed on 4 February 2021).
- Jun, J.K.; Kim, S.M. Association study of fibroblast growth factor 2 and fibroblast growth factor receptors gene polymorphism in korean ossification of the posterior longitudinal ligament patients. J. Korean Neurosurg. Soc. 2012, 52, 7–13.
- Tsukahara, S.; Miyazawa, N.; Akagawa, H.; Forejtova, S.; Pavelka, K.; Tanaka, T.; Toh, S.; Tajima, A.; Akiyama, I.; Inoue, I. COL6A1, the candidate gene for ossification of the posterior longitudinal ligament, is associated with diffuse idiopathic skeletal hyperostosis in Japanese. Spine 2005, 30, 2321–2324.
- Roberts, S.J.; Ke, H.Z. Anabolic Strategies to Augment Bone Fracture Healing. Curr. Osteoporos. Rep. 2018, 16, 289–298.
- Moosa, S.; Wollnik, B. Altered FGF signalling in congenital craniofacial and skeletal disorders. Semin. Cell Dev. Biol. 2016, 53, 115–125.
- Jann, J.; Gascon, S.; Roux, S.; Faucheux, N. Influence of the TGF-beta Superfamily on Osteoclasts/Osteoblasts Balance in Physiological and Pathological Bone Conditions. Int. J. Mol. Sci. 2020, 21, 7597.
- Gupta, A.; Zimmermann, M.T.; Wang, H.; Broski, S.M.; Sigafoos, A.N.; Macklin, S.K.; Urrutia, R.A.; Clark, K.J.; Atwal, P.S.; Pignolo, R.J.; et al. Molecular characterization of known and novel ACVR1 variants in phenotypes of aberrant ossification. Am. J. Med. Genet. A 2019, 179, 1764–1777.
- Tsukamoto, S.; Kuratani, M.; Katagiri, T. Functional characterization of a unique mutant of ALK2, p.K400E, that is associated with a skeletal disorder, diffuse idiopathic skeletal hyperostosis. Bone 2020, 137, 115410.
- Lopez, C.D.; Bekisz, J.M.; Corciulo, C.; Mediero, A.; Coelho, P.G.; Witek, L.; Flores, R.L.; Cronstein, B.N. Local delivery of adenosine receptor agonists to promote bone regeneration and defect healing. Adv. Drug Deliv. Rev. 2019, 146, 240–247.
- Corciulo, C.; Cronstein, B.N. Signaling of the Purinergic System in the Joint. Front. Pharmacol. 2019, 10, 1591.
- Daniels, G.; Ballif, B.A.; Helias, V.; Saison, C.; Grimsley, S.; Mannessier, L.; Hustinx, H.; Lee, E.; Cartron, J.P.; Peyrard, T.; et al. Lack of the nucleoside transporter ENT1 results in the Augustine-null blood type and ectopic mineralization. Blood 2015, 125, 3651–3654.
- Dixon, J.; Beaucage, K.; Nagao, M.; Lajoie, G.; Veras, M.; Fournier, D.; Holdsworth, D.; Bailey, C.; Hammond, J.; Séguin, C. Mice lacking the nucleoside transporter ENT1: A model of diffuse idiopathic skeletal hyperostosis in humans. In Proceedings of the Orthopaedic Proceedings, Montreal, QC, Canada, 17 July 2020; p. 123.
- Strazzulla, L.C.; Cronstein, B.N. Regulation of bone and cartilage by adenosine signaling. Purinergic Signal. 2016, 12, 583–593.
- Husain, A.; Jeffries, M.A. Epigenetics and Bone Remodeling. Curr. Osteoporos. Rep. 2017, 15, 450–458.
- Chiba, N.; Furukawa, K.; Takayama, S.; Asari, T.; Chin, S.; Harada, Y.; Kumagai, G.; Wada, K.; Tanaka, T.; Ono, A.; et al. Decreased DNA methylation in the promoter region of the WNT5A and GDNF genes may promote the osteogenicity of mesenchymal stem cells from patients with ossified spinal ligaments. J. Pharmacol. Sci. 2015, 127, 467–473.
- Yuan, X.; Shi, L.; Chen, Y. Non-coding RNAs in ossification of spinal ligament. Eur. Spine J. 2021, 30, 801–808.
- Hong, L.; Sun, H.; Amendt, B.A. MicroRNA function in craniofacial bone formation, regeneration and repair. Bone 2021, 144, 115789.
- Zhang, H.; Xu, C.; Liu, Y.; Yuan, W. MicroRNA-563 promotes the osteogenic differentiation of posterior longitudinal ligament cells by inhibiting SMURF1. Zhonghua Wai Ke Za Zhi 2017, 55, 203–207.
- Esser, J.S.; Saretzki, E.; Pankratz, F.; Engert, B.; Grundmann, S.; Bode, C.; Moser, M.; Zhou, Q. Bone morphogenetic protein 4 regulates microRNAs miR-494 and miR-126-5p in control of endothelial cell function in angiogenesis. Thromb. Haemost. 2017, 117, 734–749.
- Pillai, S.; Littlejohn, G. Metabolic factors in diffuse idiopathic skeletal hyperostosis—A review of clinical data. Open Rheumatol. J. 2014, 8, 116–128.
- Chaput, C.D.; Siddiqui, M.; Rahm, M.D. Obesity and calcification of the ligaments of the spine: A comprehensive CT analysis of the entire spine in a random trauma population. Spine J. 2019, 19, 1346–1353.
- Sohn, S.; Chung, C.K.; Han, I.; Park, S.B.; Kim, H. Increased Bone Mineral Density in Cervical or Thoracic Diffuse Idiopathic Skeletal Hyperostosis (DISH): A Case-Control Study. J. Clin. Densitom. 2018, 21, 68–74.
- Mader, R.; Novofestovski, I.; Adawi, M.; Lavi, I. Metabolic syndrome and cardiovascular risk in patients with diffuse idiopathic skeletal hyperostosis. Semin. Arthritis Rheum. 2009, 38, 361–365.
- Glick, K.; Novofastovski, I.; Schwartz, N.; Mader, R. Cardiovascular disease in diffuse idiopathic skeletal hyperostosis (DISH): From theory to reality-a 10-year follow-up study. Arthritis Res. Ther. 2020, 22, 190.
- Fassio, A.; Adami, G.; Idolazzi, L.; Giollo, A.; Viapiana, O.; Bosco, E.; Negrelli, R.; Sani, E.; Sandri, D.; Mantovani, A.; et al. Diffuse Idiopathic Skeletal Hyperostosis (DISH) in Type 2 Diabetes: A New Imaging Possibility and a New Biomarker. Calcif. Tissue Int. 2021, 108, 231–239.
- Mader, R.; Lavi, I. Diabetes mellitus and hypertension as risk factors for early diffuse idiopathic skeletal hyperostosis (DISH). Osteoarthr. Cartilage 2009, 17, 825–828.
- Corkill, M.; Topless, R.; Worthington, A.; Mitchell, R.; Gregory, K.; Stamp, L.K.; Brown, M.; Merriman, T.R.; Dalbeth, N. Exploring the Relationship between Gout and Diffuse Idiopathic Skeletal Hyperostosis (DISH): An Epidemiologic and Genetic Study. Arthritis Rheumatol. 2018, 70 (Suppl. 10). Available online: (accessed on 4 February 2021).
- El Miedany, Y.M.; Wassif, G.; el Baddini, M. Diffuse idiopathic skeletal hyperostosis (DISH): Is it of vascular aetiology? Clin. Exp. Rheumatol. 2000, 18, 193–200.
- Pappone, N.; Ambrosino, P.; di Minno, M.N.D.; Iervolino, S. Is diffuse idiopathic skeletal hyperostosis a disease or a syndrome? Rheumatology 2017, 56, 1635–1636.
- Bakker, J.T.; Kuperus, J.S.; Kuijf, H.J.; Oner, F.C.; de Jong, P.A.; Verlaan, J.J. Morphological characteristics of diffuse idiopathic skeletal hyperostosis in the cervical spine. PLoS ONE 2017, 12, e0188414.

