Chemotherapy-induced peripheral neuropathy (CIPN), one of major dose-limiting side effects of first-line chemotherapeutic agents such as paclitaxel, oxaliplatin, vincristine, and bortezomib is resistant to most of existing medicines.
- high mobility group box 1 (HMGB1)
- chemotherapy-induced peripheral neuropathy (CIPN)
1. Introduction
Chemotherapy-induced peripheral neuropathy (CIPN) is one of the major dose-limiting side effects of first-line chemotherapeutic agents such as paclitaxel, oxaliplatin, vincristine, and bortezomib. In terms of evidence-based medicine, no drugs are recommended for the prevention of CIPN, and duloxetine is the only agent that has limited benefit in treating established CIPN [1]. The mechanisms for development and maintenance of CIPN have not been fully understood, although preclinical studies have provided evidence for some possible mechanisms for CIPN [2], such as neuroimmune interactions [3], mitochondrial dysfunction [4], reactive oxygen species (ROS) accumulation [5], and transcriptional or functional upregulation of cation channels [6][7] [6,7] in the spinal cord, dorsal root ganglion (DRG), and peripheral sensory neurons. Our preclinical studies have demonstrated the involvement of high mobility group box 1 (HMGB1) in the development and maintenance of CIPN [8][9][10][11][8,9,10,11]. HMGB1 (also known as amphoterin) is a non-histone nuclear protein that has highly conserved amino acid sequences during evolution and is ubiquitously expressed in most mammalian cell types [12]. HMGB1 is essential for life, because HMGB1-deficient mice die within 24 h of birth due to hypoglycemia [13]. In the nucleus, HMGB1 contributes to nucleosome stability and sliding, DNA replication and repair, gene transcription, etc. On the other hand, HMGB1 is actively secreted by activated immune cells and passively released from necrotic cells in pathological conditions including inflammation. Extracellular HMGB1 acts as a damage-associated molecular pattern (DAMP) protein via activation of several pattern recognition receptors (PRRs) including Toll-like receptors (TLRs), receptors for advance glycosylation end products (RAGE), C-X-C motif chemokine receptor 4 (CXCR4), etc., leading to acceleration of inflammation and pain [11][13][14][15][16][17][18][19][20][11,12,14,15,16,17,18,19,20]. There are plenty of papers showing that an anti-HMGB1-neutralizing antibody (HMGB1-nAb) [21][22] [21,22] strongly suppresses somatic or visceral pathological pain with inflammatory and/or neuropathic components [23][24][25][26][27][28][29][30][31][32][33][34][23,24,25,26,27,28,29,30,31,32,33,34]. We have shown that the HMGB1-nAb strongly prevents the development of CIPN in rodents [8][9][10][8,9,10]. Similarly, recombinant human soluble thrombomodulin (TM) [thrombomodulin alfa (TMα), ART-123, recomodulin®], capable of promoting thrombin-dependent HMGB1 degradation [11][35][36][11,35,36], also prevents the development of CIPN in rodent models [8][9][10][8,9,10]. Intriguingly, the efficacy of TMα in preventing CIPN in humans has been confirmed by a placebo-controlled, randomized, double-blind phase IIa study [37]. Thus, targeting extracellular HMGB1 is considered a promising strategy to prevent CIPN.
2. Role of HMGB1 in CIPN
2.1. Involvement of Endogenous HMGB1 in the Development of Pathological Pain Including CIPN
In 2001, it was reported for the first time that injection of HMGB1 around the sciatic nerve induced mechanical allodynia in rats [38]. Recently, increasing evidence has unveiled the pro-nociceptive role of HMGB1 in the peripheral tissue and spinal cord [16][18][29][39][40], and demonstrated that endogenous HMGB1 is involved in the pathogenesis of various types of intractable pain [11][15], including inflammatory pain [28][29][39], visceral pain [19][20][30][32], neuropathic pain [23][31][41][42][43], cancer pain [33], and post-stroke pain [34]. Endogenous HMGB1 also appears to play a key role in the development of CIPN in rats or mice treated with cancer chemotherapeutics, such as paclitaxel, oxaliplatin, and vincristine, considering the complete prevention of CIPN by inactivation of HMGB1 with HMGB1-nAb or TMα (
2.1. Involvement of Endogenous HMGB1 in the Development of Pathological Pain Including CIPN
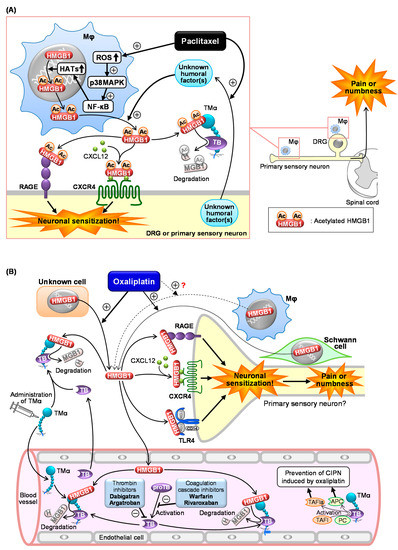
Figure 12.
A
B
A
B
2.2. Roles of Macrophage-Derived HMGB1 and Its Upstream/Downstream Molecules in CIPN Caused by Paclitaxel
Figure 1A) [9][44]. In macrophage-like RAW264.7 cells, paclitaxel causes cytoplasmic translocation and extracellular secretion of nuclear HMGB1, which involves the activation of the ROS/p38MAPK/NF-κB pathway and subsequent upregulation of histone acetyltransferases (HATs), possibly essential for acetylation of HMGB1 (
2A) [9,93]. In macrophage-like RAW264.7 cells, paclitaxel causes cytoplasmic translocation and extracellular secretion of nuclear HMGB1, which involves the activation of the ROS/p38MAPK/NF-κB pathway and subsequent upregulation of histone acetyltransferases (HATs), possibly essential for acetylation of HMGB1 (Figure 1A) [9]. Deletion of ROS by
2A) [9]. Deletion of ROS byN
-acetylcysteine (NAC), an antioxidant, prevents the development of CIPN in mice treated with paclitaxel in vivo (Table 1), in agreement with the in vitro experiments using macrophages [9]. Co-culture experiments using macrophage-like RAW264.7 cells and neuron-like NG108-15 cells suggest that “unknown humoral factors” derived from neurons accelerate HMGB1 secretion by macrophages in response to paclitaxel (Figure 1A) [9]. Thus, a crosstalk between macrophages and neurons mediated by HMGB1 and unknown humoral factors may play a key role in CIPN development after paclitaxel treatment.
2A) [9]. Thus, a crosstalk between macrophages and neurons mediated by HMGB1 and unknown humoral factors may play a key role in CIPN development after paclitaxel treatment. Antagonists of RAGE or CXCR4, membrane receptors targeted by at-HMGB1, prevent the development of CIPN caused by paclitaxel (Figure 1A) [9]. On the other hand, the role of TLR4 in the paclitaxel-induced CIPN appears to be different among species or strains. TAK-242, a TLR4 antagonist capable of penetrating into the CNS, prevents CIPN after paclitaxel treatment in C57BL/6 mice and Sprague–Dawley rats, but not in ddY mice [9][45]. It is to note that intraperitoneal (i.p.) administration of LPS-RS, a peripherally preferential TLR4 antagonist, does not prevent the CIPN in C57BL/6 mice (Table 1) [9], while intrathecal administration of LPS-RS blocks rat CIPN caused by paclitaxel [46]. Therefore, the CIPN caused by paclitaxel appears to involve central, but not peripheral, TLR4 in rats and C57BL/6 mice. Interestingly, there is a report showing that macrophage TLR9, an intracellular compartment receptor for HMGB1, participates in the CIPN caused by paclitaxel in male, but not female, mice, suggesting the existence of sex dimorphism in the role of TLR9 in the development of CIPN [47]. The same study has shown the involvement of TLR9 in the paclitaxel-induced release of TNF and CXCL1 from male, but not female, macrophages [47]. Similar sex dimorphisms have also been described in the role of TLR4 in pain processing [39][48]. Collectively, activation of RAGE and CXCR4 possibly by macrophage-derived at-HMGB1 may play a major part in CIPN due to paclitaxel.
2A and Table 1) [9]. On the other hand, the role of TLR4 in the paclitaxel-induced CIPN appears to be different among species or strains. TAK-242, a TLR4 antagonist capable of penetrating into the CNS, prevents CIPN after paclitaxel treatment in C57BL/6 mice and Sprague–Dawley rats, but not in ddY mice (Table 1) [9,94]. It is to note that intraperitoneal (i.p.) administration of LPS-RS, a peripherally preferential TLR4 antagonist, does not prevent the CIPN in C57BL/6 mice (Table 1) [9], while intrathecal administration of LPS-RS blocks rat CIPN caused by paclitaxel (Table 1) [95]. Therefore, the CIPN caused by paclitaxel appears to involve central, but not peripheral, TLR4 in rats and C57BL/6 mice. Interestingly, there is a report showing that macrophage TLR9, an intracellular compartment receptor for HMGB1, participates in the CIPN caused by paclitaxel in male, but not female, mice, suggesting the existence of sex dimorphism in the role of TLR9 in the development of CIPN [96]. The same study has shown the involvement of TLR9 in the paclitaxel-induced release of TNF and CXCL1 from male, but not female, macrophages [96]. Similar sex dimorphisms have also been described in the role of TLR4 in pain processing [80,97]. Collectively, activation of RAGE and CXCR4 possibly by macrophage-derived at-HMGB1 may play a major part in CIPN due to paclitaxel.2.3. Role of HMGB1 and Its Upstream/Downstream Molecules in CIPN Caused by Oxaliplatin
We have shown that the inactivation of HMGB1 with HMGB1-nAb or TMα and pharmacological blockade of RAGE, CXCR4, or TLR4 prevent CIPN in rodents treated with oxaliplatin (Figure 2B) [10]. Peripheral TLR4 appears to be involved in the CIPN caused by oxaliplatin, because systemic administration of LPS-RS as well as TAK-242 exhibited a preventive effect in this CIPN model (Figure 12B). Most surprisingly, macrophages do not play a major role in the CIPN caused by oxaliplatin, since depletion of macrophages by liposomal clodronate as well as inhibition of macrophage-derived HMGB1 by minocycline or ethyl pyruvate had no effect on the CIPN in mice treated with oxaliplatin [10]. An independent group [49] [98] has suggested the involvement of macrophages in the same CIPN model, being apparently inconsistent with our results [10]. However, their study showed that liposomal clodronate only slightly (by 30%) reduced the oxaliplatin-induced allodynia, suggesting that the participation of macrophage-derived HMGB1 in the CIPN caused by oxaliplatin should be very minor, if any [50][99]. This notion is further supported by our findings that the number of macrophages in the sciatic nerve did not increase in mice treated with oxaliplatin. Thus, the origin of HMGB1 involved in CIPN caused by oxaliplatin is still open to question (Figure 12B). Alternatively, it is likely that HMGB1 derived from multiple cells including macrophages contributes to the development of CIPN following oxaliplatin treatment (Figure 12B), because oxaliplatin at relatively high concentrations (3–10 µM) causes HMGB1 release from Schwann cells and macrophage-like RAW264.7 cells in vitro [10]. Our study to explore other origins that secrete HMGB1 in response to oxaliplatin is now in progress.

