Obesity is a modern health problem that has reached pandemic proportions. It is an established risk factor for carcinogenesis, however, evidence for the contribution of adipose tissue to the metastatic behavior of tumors is also mounting. Over 90% of cancer mortality is attributed to metastasis and metastatic tumor cells must communicate with their microenvironment for survival. Many of the characteristics observed in obese adipose tissue strongly mirror the tumor microenvironment. Thus in the case of prostate, pancreatic and breast cancer and esophageal adenocarcinoma, which are all located in close anatomical proximity to an adipose tissue depot, the adjacent fat provides an ideal microenvironment to enhance tumor growth, progression and metastasis. Adipocytes provide adipokines, fatty acids and other soluble factors to tumor cells whilst immune cells infiltrate the tumor microenvironment.
- obesity
- adipose tissue
- tumor progression
- cancer relapse
- metastasis
- cytokines
- adipokines
- extracellular matrix
- extra cellular vesicles
- cancer metabolism
1. Introduction
The metabolic and cardiovascular risks of obesity are well known. However, it is estimated that 40% of all cancer deaths are also attributable to obesity [1]. Indeed, globally, excess body weight is third behind smoking and infection as an attributable risk factor for cancer, and second to smoking in Western populations [2]. Obesity adversely effects cancer in two ways, (i) by promoting carcinogenesis resulting in a higher cancer incidence and (ii) cancer progression resulting in an increased risk of mortality [3]. In breast cancer, for example, obesity is only associated with an increased incidence of post-menopausal breast cancer, whilst obesity is a risk factor for progression in all breast cancer subtypes [4]. The global obesity rate in women is projected to reach 21% by 2025 and this is particularly alarming considering that 55% of all female cancers have an obesity associated mechanism [3]. Central obesity, resulting from the overgrowth of visceral white adipose tissue (WAT), has been specifically linked to cancer progression [5]. While diet is undoubtedly important in obesity, animal models have indicated that WAT overgrowth directly promotes cancer progression irrespective of diet [3,6][3][6]. Epidemiological studies have demonstrated a compelling association between cancer risk and obesity. Analysis of the association between body mass index (BMI) and early stage breast cancer outcomes in the Danish Breast Cancer Cooperative Group (n = 53,816 women) revealed that obese women have a 46% higher risk of developing distinct metastasis at 10 year follow up compared to normal weight women [7]. Furthermore, a meta-analysis of 82 studies found a 41% and 35% higher risk, respectively, of all-cause mortality and breast cancer specific mortality in obese women compared to normal weight women [2]. An umbrella review of 204 meta-analyses revealed a strong association between obesity and gastrointestinal cancers including esophageal adenocarcinoma (OAC) [8]. OAC was notable for a progressive increase in risk ratio (RR) for each 5 kg/m2 increase in BMI (RR 4.8 for a BMI ≥ 40 kg/m2) suggesting a dose–response effect [9]. The top two cancers demonstrating very strong associations with BMI are endometrial (RR 1.48) and OAC (RR 1.45) [10].
In obesity, white adipocytes become hypertrophic and hyperplasic, which results in physiologic changes, including elevated free fatty acids (FFAs) and triglycerides, increased blood glucose and insulin resistance. Obese adipose tissue increases production of proinflammatory cytokines, e.g., tumor necrosis factor (TNFα), interleukin-6 (IL-6), interleukin-1β (IL-1β) and adipokines (e.g., leptin) [3]. Metastasis is the primary cause of cancer morbidity and mortality and efforts to unravel the molecular mechanisms linking dysfunctional adipose tissue and the ability of tumor cells to acquire metastatic properties will lead to the discovery of novel targets for metastasis.
2. The Metastatic Cascade
The “"hallmarks of cancer”" define characteristics that are critical for cellular transformation [11]. Among the hallmarks there is only one defining factor, invasion, which distinguishes a malignant and benign tumor [11]. The metastatic cascade begins with local invasion before progressing to intravasation, arrest at distant organs, extravasation, micro metastasis formation and finally metastatic outgrowth (Figure 1).
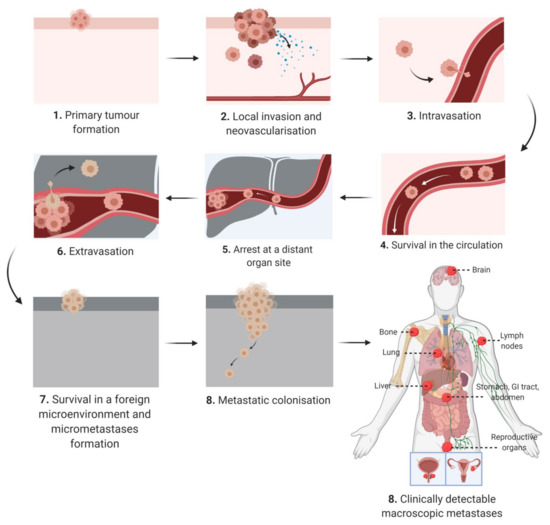
Schematic diagram depicting the keys steps of the metastatic cascade from initial presentation as an in situ tumor mass at the primary site to macroscopically detected metastatic lesions at secondary sites.
In the early stages of tumor development, the primary tumor cell mass typically has an expansive phase in the absence of invasion, encapsulated in a dense fibrous network (i.e., desmoplasia) [12]. A subset of neoplastic cells acquire the ability to escape through the basement membrane and detach from the primary tumor [12]. The dissemination of cancer cells is a consequence of chromosomal instability that causes continuous errors in chromosome segregation during mitosis [13]. This in turns leads to the rupture of micronuclei and the secretion of genomic DNA into the cytosol, which activates DNA sensing pathways and NF-κB signaling [13]. In addition, epithelial–mesenchymal transition (EMT) is a transdifferentiating process, which permits epithelial cells to attain a mesenchymal phenotype with migratory potential [14]. Spontaneous EMT in primary tumor cells can be triggered by hypoxia, metabolic stressors and matrix stiffness [15,16][15][16]. Although some studies have cast doubts on the necessity of EMT during metastasis [17[17][18],18], there is evidence for cells expressing both epithelial and mesenchymal markers within the primary tumor, circulation and at a secondary metastatic site [19]. Therefore EMT is being increasingly understood as a spectrum of transitional stages between epithelial and mesenchymal phenotypes rather than a binary choice between an epithelial or mesenchymal phenotype [20,21][20][21]. There is evidence that cells in different EMT stages prefer certain microenvironments, for example, metastatic cells with a predominating mesenchymal phenotype proliferate near endothelial and inflammatory cells [21]. These tumor cells release factors to attract immune cells and stimulate angiogenesis, thus promoting an inflammatory and highly vascularized niche [21]. Although EMT is required for metastatic dissemination, the opposite process of mesenchymal to epithelial transition (MET) is required for metastatic colonization of distant sites [22]. There is also substantial evidence that disseminated tumor cells express stem cell markers, such as aldehyde dehydrogenase (ALDH), and functionally these cells, highly enriched in stem cells markers, have an enhanced ability to cause metastasis [19,23,24,25][19][23][24][25]. Furthermore, genome wide analysis of both cells undergoing EMT and circulating tumor cells has revealed a similar transcriptome to primary cancer stem cells (CSCs) thus indicating an overlapping subpopulation [19,26,27][19][26][27]. In pancreatic cancer, primary CD133+ CSCs demonstrated classic CSC characteristics such as tumor initiation and chemoresistance [28]. However, at the invasive front of the tumor CD133+ cells are enriched for CXC-chemokine receptor 4 (CXCR4) and the CD133+CXCR4+ population is more migratory than CD133+CXCR4-- [28]. Moreover, patients with increased CD133+CXCR4+ cells had more metastatic disease [28]. This indicates that microenvironmental cues within the tumor can trigger heterogeneity in CSCs and CD133+CXCR4+ and CD133+CXCR4- are not a distinct subpopulations but a gradient of stemness phenotypes [19,28][19][28]. CSCs are more resistant to chemotherapy due to higher expression of multidrug resistance (MDR) or detoxification proteins such as aldehyde dehydrogenase (ALDH) [29].
Prior to exiting the primary tumor mass, tumor cells communicate with other microenvironments, termed the premetastatic niche, and this niche is selectively primed by secreted factors and extracellular vesicles to induce vascular leakage, extra cellular matrix (ECM) remodeling and immunosuppression [30]. Cancer cells also adjust the niche themselves by remodeling the ECM leading to stromal tumorigenesis [20]. Cancer patients release large numbers of cancer cells into the circulation daily, however animal studies of melanoma suggest that <0.1% of these cells metastasize [31]. The most widely studied route of dissemination is through the bloodstream (hematogenous), however metastatic cells may also migrate along nerves, lymphatic vessels, across coelomic cavities or along the basal side of endothelial cells and never enter the lumen [12]. Intravasation, the dissemination of cancer cells to organs through the lumen of the vasculature can be active or passive depending on the tumor type, microenvironment and vasculature [32]. The migration of metastatic cells into the circulation relies on chemokines and complement components that direct tumor cells through the vasculature and metabolic factors that result in an antioxidant effect [20]. Furthermore, the dissemination of circulating tumor cells (CTCs) is supported by a close association with immune cells such as activated platelets, macrophages and neutrophils [20]. When CTCs pass into small capillaries they become trapped leading to microvascular rupture or the cell undergoes extravasation [20]. Establishing a vascular network is required for metastatic colonization and this can occur though angiogenesis, co-opting existing vessels or inducing vasculogenic mimicry [20,33][20][33]. Furthermore, cancer cells can also exploit neuronal signaling pathways for growth and adaptation [20]. Cancer dormancy is an arrest phase that can occur after invasion into secondary sites and in some cancer survivors, dormant cells result in relapse long after the removal of the primary tumor [20,34][20][34]. The subsequent reactivation is governed by self-renewal pathways (e.g., Wnt, Hedgehog and Notch) and cells exhibit increased levels of stem cell associated genes [35]. While dormant cancer cells downregulate the expression of immune cell recognizable antigens, persistent host organ inflammation and the establishment of neutrophils in extracellular traps may transform dormant cells into aggressive metastasis [35,36][35][36]. In many of the steps from dissemination to colonization, the microenvironment plays a major role and this can be significantly altered by dysfunctional adipose tissue in people living with obesity.
3. Mechanisms Linking Adipose Tissue to the Metastatic Cascade
3.1. Adipocytes and Adipokines
Adipocytes secrete more than 600 soluble factors, known as adipokines, and the most well characterized are leptin and adiponectin [37]. Intra-abdominal cancers such as ovarian, colon and gastric, preferentially metastasize to the omentum, a peritoneal organ largely composed of adipocytes, suggesting that adipocytes significantly contribute to the metastatic cascade [38]. Omental adipocytes promote migration and invasion of ovarian cancer cells by secreting cytokines [39]. Neutralization of these cytokines reduced in vivo homing of ovarian cancer cells to mouse omentum, suggesting that adipocytes promote the early stages of metastasis [39]. In addition, ovarian cancer cells metabolically adapt to the increased availability of lipids by utilizing energy from fatty acids for growth [39]. Fatty acid binding protein 4 (FABP4) was strongly expressed at the adipocyte–cancer cell interface and pharmacological inactivation of FABP4 decreased cancer cell lipid accumulation, invasiveness and omental metastasis [39]. Circulating levels of FABP4 are markedly increased in obese individuals due to release from an expanded adipose tissue depot and FABP4 can induce mammary tumor stem cells by enhancing ALDH1 activity via IL-6/STAT3 signaling [40,41][40][41]. In addition, FABP4 promoted aggressive acute myeloid leukemia (AML) in obesity by enhancing aberrant DNA methyltransferase 1 (DNMT1)-dependent DNA methylation of tumor cells [42]. Upon interaction with cancer cells, adipocytes dedifferentiate into preadipocytes or are reprogrammed into cancer-associated adipocytes (CAAs), which resemble fibroblasts and have dispersed lipid droplets [43]. In breast cancer, matrix metalloproteinase (MMP)-11 is induced in adipocytes by adjacent invading cancer cells. In the presence of MMP-11, the activated adipocyte dedifferentiates into a preadipocyte fibroblast-like cell, which can sustain cancer cell invasion [44,45][44][45]. Bone marrow adipocytes constitute approximately 15% of bone marrow volume in young adults, rising to 60% by the age of 65 years old [46]. They have a distinctive phenotype, which resembles both white and brown adipose tissue, and they secrete fatty acids, cytokines and adipokines, which influence the whole bone microenvironment [47]. The bone provides a supportive microenvironment for both solid tumor and hematological metastasis, including breast, prostate and multiple myeloma and bone metastatic cancers primarily occur in older adults whose bone marrow is heavily populated by adipocytes [48,49][48][49]. Similarly to the omentum, cancer cells are attracted to the adipocytes in the metabolically active red marrow and this creates a niche in the bone marrow for disseminated cancer cells to establish and progress [48]. In addition, leukemic stem cells expressing the fatty acid transporter CD36, induce lipolysis in gonadal adipose tissue to support their metabolism and evade chemotherapy [50]. Lipids can also be trafficked between bone marrow adipocytes and cancer cells by upregulation of FABP4 and fuel growth and invasiveness in metastatic tumor cells [51]. In addition, bone marrow adipocytes have been shown to promote the Warburg phenotype in metastatic prostate cancer cells through oxygen independent HIF-1α activation [52]. Bone marrow adipocytes are a major source of circulating adiponectin, much greater than WAT [53]. Adiponectin is reported to suppress many elements of the early metastatic cascade including adhesion, invasion, migration and stem cell properties via numerous signaling pathways including WNT, NF-κB and JAK/STAT [54]. In advanced cancer associated with cachexia, hyperadiponectinemia has been observed [55]. In addition, increased adiponectin signaling in dendritic cells can blunt anti-tumor immune responses in patients with metastatic disease [56]. However, the late increase in adiponectin has very little influence on the course of the disease, as its role is thought to be more prominent in early metastatic spread [54]. During the development of obesity, preadipocytes differentiate incorrectly leading to hypoxia and the induction of hypoxia induced factor-1 (HIF-1) [57]. This inhibits the expression of adiponectin and increases the expression of leptin, resulting in a reduced adiponectin to leptin ratio in obesity-associated adipose tissue [58,59][58][59]. A high leptin to adiponectin ratio has been reported to increase the risk of postmenopausal and triple negative breast cancer (TNBC) progression [60,61][60][61]. Leptin is another adipokine important in tumor progression and secretion of leptin is increased in CAAs compared to mature adipocytes [43]. Leptin levels are increased in the plasma of post-menopausal breast cancer patients, which correlated with a higher grade, advanced tumor stages and presence of distant metastasis [37]. Leptin exerts it effect through the transmembrane leptin receptor and only the full length isoform, ObRb, contains the intracellular domain required for JAK/STAT signaling [62]. ObRb was significantly overexpressed in metastatic lymph nodes compared to the primary tumor in ER-breast cancer patients [63]. Furthermore, TNBC patient derived xenografts (PDX) grown in the presence of primary adipose stem cells (ASCs) isolated from obese donors (obASCs) had increased HLA1+ human tumor cells and CD44+CD24− CSCs in the peripheral blood and metastasis compared to ASCs from lean women [64]. In addition, the knockdown of leptin expression in obASCs suppressed the prometastatic effect [64]. There is a plethora of evidence that leptin induces cell migration and invasion in breast cancer via JAK/STAT3 signaling [62]. In addition, the Notch signaling pathway is a key regulator of leptin induced cell migration in breast cancer obesity models [63]. Leptin has also been shown to promote the CSC phenotype though STAT3 activation and this in turn recruited a histone methyl transferase causing a repression of miR-200c by epigenetic silencing and the expansion of CSCs [65]. Autotaxin (ATX) is an adipocyte-derived lysophospholipase D that catalyzes the hydrolysis of circulating or cell associated lysophosphatidylcholine (LPC) to the bioactive lipid lysophosphatidic acid (LPA) [66]. Some tumor cells, such as in melanoma, glioblastoma and thyroid cancer, directly secrete ATX leading to a chronic inflammatory state and decreased acquired immune response [67]. In contrast, other tumor cells, such as breast, ovarian and pancreatic do not produce ATX and instead the tumor microenvironment is the primary source [67]. Interestingly, the ATX gene (ENPP2) was the second most upregulated gene in breast CSCs treated with paclitaxel whilst the LPP2 gene (PLPP2) was downregulated, indicating CSCs may favor an LPA-enriched microenvironment [68]. In a tissue microarray (TMA) of metastatic breast cancer, stromal ATX was highly expressed in bone metastasis [69]. In mice approximately 40% of ATX is produced by adipocytes and this increases when mice are fed a high fat diet [70]. Similarly, in humans ATX production increases in obesity, particularly in inflamed adipose tissue, and contributes to comorbidities including insulin resistance, hepatic steatosis and atherosclerosis [66,71,72,73][66][71][72][73]. In breast cancer cells both ATX and LPA are associated with mobility and invasive capacity via the JAK/STAT3 pathway or PI3K/MAPK pathways [74,75][74][75]. Therefore cross talk between adipose tissue derived ATX and tumors cells is a potential mechanism for tumor progression [76,77][76][77]. Indeed preclinical studies of an ATX inhibitor, ONO-843050 suppresses tumor growth and 60% of lung metastasis in a breast cancer mouse model [78]. Targeting other molecules in the ATX-LPA signaling pathways also results in decreased breast cancer metastasis formation in murine models [77,79,80][77][79][80]. The physiological upregulation of ATX occurs in response to inflammation and chronic activation of ATX-LPA signaling occurs in diseases such as pulmonary fibrosis, rheumatoid arthritis and inflammatory bowel diseases [81]. A first in class ATX inhibitor, GLPG1690, attenuated idiopathic pulmonary fibrosis in a Phase IIa clinical trial (NCT02738801) and two Phase III clinical trials are currently underway for this indication (NCT03711162; NCT03733444) [82,83][82][83]. In a preclinical breast cancer model, GLPG1690 acted synergistically with chemotherapy and radiotherapy to improve outcomes [84]. Considering the role of ATX in regulating CSCs and mobility, targeting ATX with inhibitors such as GLPG1690 may also prove to be beneficial in targeting metastasis, particularly in patients living with obesity, by preventing adipose tissue cross talk.
3.2. Immune Cells and Inflammatory Factors
CAAs secrete more inflammatory factors, such as monocyte chemoattractant protein (MCP-1), RANTES, IL-1β, IL-6 and TNFα than “"normal”" adipocytes and this can promote invasion and metastasis formation [43,85][43][85]. Furthermore, the recruitment and activation of immune cell subsets, particularly M1 macrophages, in obese WAT increases local and systemic levels of proinflammatory cytokines [86]. MCP-1 is elevated in obesity and secreted by many cells including tumor cells, fibroblasts, tumor infiltrating monocytes and endothelial cells [43,87][43][87]. In cancer cells, MCP-1 induces the expression of NOTCH1 and subsequently promotes the activity of CSCs and neovascularization [87,88][87][88]. RANTES expression in the peritumoral adipose tissue of women with TNBC correlated with lymph node and distant metastasis [89]. Furthermore, the RANTES inhibitors, maraviroc and vicriciroc, reduced invasion and pulmonary metastasis in a preclinical tumor model of breast cancer [90]. IL-6 is a pleotropic cytokine involved in immune regulation and tumorigenesis. One third of total circulating IL-6 originates from adipocytes and circulating levels are correlated with adiposity [91,92][91][92]. When adipocytes are cocultured with breast cancer cells, adipocytes increase secretion of IL-6, which in turn promotes invasion and migration of tumor cells [93]. Furthermore, IL-6 also plays a critical role in the biology of CSCs through activation of Notch and JAK/STAT signaling [94,95][94][95]. In addition to classical IL-6 signaling, IL-6 trans signaling is a major driver of obesity associated hepatocellular carcinoma (HCC), through inhibition of p53 induced apoptosis and enhanced angiogenesis [96]. IL-6 also promoted HCC progression via upregulation of osteopontin (OPN), a secretory ECM protein involved in the maintenance of the stemness phenotype [97].
The inflammasome is a highly regulated protein complex that triggers caspase-1 activation and subsequent secretion of IL-1β and IL-18 [98]. Obesity related factors, such as cholesterol crystals and fatty acids (palmitate and ceramide), can lead to unchecked activation of the inflammasome [98]. IL-1β expression in primary tumors is a potential biomarker for predicting breast cancer patients who are at increased risk of developing bone metastasis [99,100][99][100]. Furthermore, in vitro studies indicate that tumor cells expressing high levels of IL-1β specifically home to and colonize the bone [101]. A persistent increase of circulating levels of TNFα occurs in obesity, mainly due to the elevated number of M1 macrophages in obese WAT [86]. TNFα stimulates the secretion of MMPs in epithelial tumors and enhances EMT to promote invasion and migration of tumor cells [85,102,103][85][102][103]. Furthermore, TNFα increases liver metastasis through inducing expression of cell adhesion molecules, such as ICAM-1, E-selectin and VCAM-1, on liver specific endothelial cells, and thus enhancing tumor cell arrest and transendothelial migration [104,105][104][105].
Crown like structures (CLSs) are a histological feature of dead or dying adipocytes surrounded by macrophages and they are increased in obesity associated adipose tissue [37]. CLS are associated with free fatty acid (FFA) release, NF-κB activation and the generation of a proinflammatory microenvironment [106]. They are best characterized for their role in the initiation and progression of breast cancer [106] but they are also reported to play a role in endometrial cancer [107], prostate cancer [108] and non-alcoholic steatohepatitis (NASH)—a major risk factor for HCC [109]. In breast adipose tissue CLS are not only associated with inflammation but they also drive aromatase activity resulting in an increased ratio of estrogen:androgen in blood and local tissues [110]. The expression of programmed death-ligand 1 (PD-L1) in adipocytes prevents the antitumor function of cytotoxic CD8+ T cells [43]. It is therefore not surprising that treatment with anti PD-1/PD-L1 immunotherapy in patients with a BMI > 25 (i.e., overweight/obese) have improved clinical outcomes compared to normal weight patients [111].
CSCs will only proliferate in specific tumor environments indicating that environmental stimuli are critical to preserve their phenotypic plasticity, to protect them from the immune system and to facilitate metastatic potential [112]. High levels of proinflammatory cytokines from obese adipose tissue can stimulate CSC properties [113]. TNFα enhances the CSC phenotype in numerous cell types and is associated with upregulation of stem cell related genes, chemo resistance and tumorigenesis [114,115,116][114][115][116]. Furthermore, TNFα upregulates TAZ (a Hippo pathway effector) and Slug (an EMT mediator), which increase breast CSCs through both canonical and non-canonical NF-κB signaling [117,118][117][118]. Analysis of TNBC patient datasets reveals high tumor expression of the epigenetic reader methyl-CpG-binding domain protein 2 (MBD2), specifically the alternative splicing variant 2 (MBD2_v2) expression and high relapse rate and high BMI [119]. It is postulated that obesity drives high reactive oxygen species (ROS) levels, which subsequently promotes MBD2_v2 expression and an expansion of the CSC fraction [119].
3.3. Angiogenesis
The processes of angiogenesis and adipogenesis are closely linked. When preadipocytes differentiate into adipocytes and become adipose tissue, new blood vessels are also formed [120]. Conversely inhibition of adipocyte differentiation also reduces angiogenesis [121]. In breast cancer, leptin upregulates all components of the IL-1 system (IL-1α, IL-1β, IL-1Ra and IL-1R tI) and both leptin and IL-1 together promote angiogenesis through expression of VEGF/VEGFR [122]. Furthermore, leptin upregulation of VEGF/VEGFR2 was impaired by IL-1 signal blockage [122]. In addition, obesity leads to an increase in tumor infiltrating macrophages with activated NLRC4 inflammasome and increased IL-1β production in breast tumors [123]. This leads to a NLRC4/IL-1β dependent upregulation of adipocyte derived angiopoietin-like 4 and enhanced obesity associated tumor angiogenesis [123]. Anti VEGF therapy has fallen short of expectations, particularly in breast cancer where the FDA revoked approval for bevacizumab because of a lack of overall survival benefit [124]. Anti VEGF therapy resistance is partly driven through expression of proinflammatory and other alternative angiogenic factors, many of which are also increased in obesity [121,125][121][125]. Furthermore, breast cancer patients with obesity are less sensitive to anti VEGF treatment and they have increased systemic concentrations of IL-6 and fibroblast growth factor-2 (FGF-2) [126]. The elevated IL-6 was associated with increased IL-6 production from adipocytes and myeloid cells within tumors and IL-6 blockage abrogated obesity induced resistance to anti VEGF therapy at both primary and metastatic sites [126]. Vasculogenic mimicry (VM) is a tumor vascular system that is independent of angiogenesis of endothelial cells, and is associated with both poor survival in multiple tumor types and anti VEGF therapy resistance [127]. Notably, the adipose tissue secretome has been shown to induce melanoma cells to arrange in 3D vessel like structures, characteristic of vasculogenic mimicry [128], supporting a role of adipose tissue in this process.
3.4. Metabolic Repogramming
An essential function of adipocytes is energy mobilization and therefore a metabolic interaction between cancer cells and adipocytes is not surprising. The Warburg effect suggests that due to mitochondrial dysfunction, malignant cells prefer to produce adenosine triphosphate (ATP) via glycolysis instead of oxidative phosphorylation, even in the presence of oxygen [129]. In parallel, cancer cells are able to use alternative sources of energy such as amino acids and lactate from the microenvironment. Bone marrow adipocytes promoted the Warburg phenotype by increased expression of glycolytic enzymes, increased lactate production and decreased mitochondrial oxidative phosphorylation in metastatic prostate cells by paracrine signaling [52]. The “"reverse Warburg effect”" theory proposes that cancer cells induce oxidative stress in the neighboring stromal cells by secreting ROS and triggering aerobic glycolysis and production of high energy metabolites, especially lactate and pyruvate. These metabolites are then transported through the “"lactate shuttle”" to sustain the anabolic needs of adjacent cancer cells [130,131][130][131]. The effect has been mainly described in stromal fibroblast cells, however given that ASCs and CAA are fibroblast like cells, it is likely they are also important contributors. Indeed it has been reported that the “"reverse Warburg effect”" was induced during the co-culture of adrenocortical carcinoma cells with ASCs [132]. Ketone bodies are another catabolite produced and released by glycolytic adipocytes and they are an ideal substrate for ATP production by driving oxidative mitochondrial metabolism leading to enhanced tumor invasiveness [91,133][91][133].
A key characteristic of CAA is their loss of lipid content. The FFAs released by adipocytes after lipolysis are stored in tumor cells as triglycerides in lipid droplets [134]. Tumor cells then release FFAs from lipid droplets though an adipose triglyceride lipase dependent lipolysis (ATGL) pathway [134]. ATGL is upregulated in tumors on contact with adipocytes and it correlates with aggressiveness by stimulating tumor cell invasion [134]. The FFAs also act as structural units for newly synthesized membrane phospholipids and cancer cell membranes become enriched with saturated and/or mono unsaturated fats leading to changes in membrane dynamics [135]. This results in cancer cells that are more resistant to oxidative induced cell death and reduced the uptake of drugs [135][134]. Advanced metastatic melanomas frequently grow in subcutaneous tissues largely composed of adipocytes [136]. Adipocyte derived lipids are transferred to melanoma through the lipid transporter FATP1 and a small molecule inhibitor of FATPs reduced melanoma growth and invasion [136]. Furthermore in AML, leukemic blasts activate lipolysis in neighboring bone marrow adipocytes leading to the transfer of lipids to the blast through FABP4 [137].
Amino acids such as glutamine, glycerine, serine and proline also have important roles in the asymmetric metabolism of amino acids between cancer and stromal cells [43]. Glutamine is a pivotal source of the TCA cycle intermediates and ATP in cancer cells, and the substrate of the antioxidant glutathione [138,139][138][139]. Stromal cells within the tumor microenvironment harness carbon and nitrogen from non-canonical sources to synthesize glutamine and it is used by the tumor cells to promote growth and metastasis [140]. Glutamine is downregulated in obesity and is inversely associated with proinflammatory gene expression and macrophage infiltration [141]. Pancreatic ductal adenocarcinoma (PDAC) cells rely on glutamine utilization to fulfill their metabolic requirements and it is the most depleted amino acid within the PDAC microenvironment [142]. Glutamine deficiency leads to the induction of EMT through the upregulation of the master EMT regulator Slug [142][136]. In addition, as glutamine levels decline, tumor cells become more reliant on asparagine for proliferation and protein synthesis [143]. Asparagine affects the metastatic cascade at multiple stages. At the primary tumor level, asparagine promotes EMT and intravasation [144,145][144][145]. In the circulation asparagine helps circulating tumor cells survive shear and oxidative stress whilst at a distinct metastatic sites, asparagine facilitates cell growth and survival by inducing glutamine synthetase (GLUL) expression and glutamine biosynthesis [144,145][144][145].
Citrulline and nitric oxide are generated by tumor cells by catabolizing the semi essential amino acid arginine [146]. Nitric oxide facilitates glycolytic activity and suppresses oxidative phosphorylation to promote proliferation [146]. Citrulline is secreted into the ECM and is taken up by stromal adipocytes, before being converted back into arginine and released for cancer cells [146]. Depriving tumor cells of arginine has cytotoxic effects through apoptosis or autophagy depending upon the tumor type, and decreasing the ability of tumor cells to migrate and adhere to the ECM [147]. Arginine dependent migration requires arginine to be metabolized by two major enzymes, arginase (ARG1) and nitric oxide synthase (NOS) [147]. In HCC, higher expression of ARG1 is positively correlated to aggressive tumor growth and poor disease free survival [148]. In vitro studies revealed that overexpression of ARG1 enhanced arginase activity leading to multiple processes that contribute to progression including increased cell viability, migration, invasion and EMT [148]. Obesity coupled with PDAC results in accelerated tumor growth and enrichment in pathways regulating nitrogen metabolism. The mitochondrial form of arginase (ARG2) that hydrolyzes arginine into ornithine and urea is induced upon obesity and is accompanied by PDAC growth and increased nitrogen flux from 15N-glutamaine into the urea cycle, the principle pathway for ammonia detoxification [149]. Infusion of 15N-arginine in murine models demonstrates a shunting of arginine catabolism away from the urea cycle into creatine synthesis, resulting in ammonia accumulation specifically in obese tumors [149]. The biological consequences of ammonia accumulation in the tumor microenvironment is not fully understood but it has been shown to directly generate amino acids through glutamate dehydrogenase activity [150].
Hyperinsulinemia is a hallmark of chronic obesity and insulin stimulates the hepatic synthesis of the peptide IGF-1 [151]. Three quarters of breast cancer patients show activation of insulin/IGF-1 signaling and this rises to 87% in patients with invasive breast cancer [152]. In preclinical models, blocking IGF in combination with paclitaxel significantly reduced tumor cell proliferation and lung metastasis [152]. Due to the frequent dysregulation of the IGF system in cancer, various components of this system became attractive anticancer targets. However, clinical trials using IGF-1 receptor blocking antibodies failed to meet expectations, except in a small number of malignancies [153,154][153][154]. More recent developments reveal that dysregulation of the IGF system results in a substantial expansion of the cancer stem like subpopulation by supporting EMT and self-renewal signaling pathways [153]. IGF signaling regulates these pathways in multiple ways though i) stimulation of the transcription factors of the ZEB and the Snail family implicated in the EMT program, ii) interacting with pluripotency transcription factors (e.g., Oct-4, SOX2, Nanog, p53 and HMGA1 proteins) and iii) regulation of development signaling factors (e.g., Wnt/β-catenin, Notch and Shh pathways) classically involved in cell stemness [153]. Intriguingly, in HER2+ breast cancer patients, high IGF-1 in normal weight patients showed a superior recurrence free survival compared to low IGF-1 [155]. In contrast, high IGF-1 in overweight patients was associated with a reduced recurrence free survival [155][153]. Obese mice have a heightened inflammatory response in the liver and an increased incidence of metastatic colon carcinoma cells to the liver [156]. Moreover, liver inflammation induced by obesity was abrogated in liver specific IGF-I deficient mice leading to a significant reduction of in liver metastasis [156]. Furthermore, IGF-1 promotes neutrophil polarization to a tumor promoting phenotype and the induction of a prometastatic microenvironment in the liver [157].
3.5. Extracellular Matrix
In addition to adipocyte hypertrophy and dysregulated lipid metabolism, heightened inflammation, hypoxia and abnormal angiogenesis, obesity is also associated with ECM remodeling. Once a primary tumor is established, cells migrate and invade to form satellite tumors within centimeters of the original tumor mass and/or disseminate through the lymph nodes and vasculature to form secondary macrometastases at distance sites (Figure 1). This progression sequence is dependent on the ability of cancer cells to traverse the ECM. The adipose tissue ECM is a three-dimensional, non-cellular structural support of the numerous cell types that reside in the adipose tissue. It is composed primarily of collagens, fibronectin and to a lesser extent lamins that are supplied by a number of resident cell types including fibroblasts, adipocytes and preadipocytes [158]. In obesity, the highly dynamic adipose tissue ECM is constantly undergoing remodeling and reorganization to accommodate increased adipocyte numbers and adipocyte hypertrophy. A rapid increase in adipose tissue volume can result in regional hypoxia, which triggers excess deposition of fibrillar collagens by adipocytes and myofibroblasts, immune cell infiltration, adipose tissue fibrosis, a desmoplastic stroma and increased tissue stiffness, with overall behavior described as similar to “"a wound that never heals”". This state of chronic low-grade inflammation within the adipose tissue drives obesity-associated pathologies including diabetes [159[159][160],160], cardiovascular disease [161] and cancer. Indeed, it is very similar to the microenvironment of a thriving tumor mass and thus trophic cancer cells that home to the adipose tissue are well supported by these suitable surroundings. Fibrosis is a hallmark of cancer, and desmoplasia within the tumor microenvironment, is a marker of poor prognosis in cancer [162,163][162][163] and can negatively impact drug delivery [164,165][164][165]. In the case of PDAC, obesity is associated with aggressive tumors with poor prognosis, and adipocyte accumulation in the malignant pancreas [166]. Obesity-induced accumulation of high adipocyte numbers in the pancreas has been shown to induce inflammation and excessive accumulation of ECM components, i.e., desmoplasia, which promotes tumor progression and resistance to chemotherapy [164].
Adipose tissue is the main component of the breast cancer microenvironment, crosstalk between the breast cancer cells and adipocytes or other adipose stromal cells stimulates the secretion of even larger quantities of ECM proteins, increasing matrix stiffness and scar formation, further enhancing EMT and local invasion of tumor cells. Within the adipose tissue, invading breast cancer cells manipulate adipocytes to form fibroblastic CAAs that secrete large volumes of ECM proteins including collagen I, III and IV, and the cleavage product of collagen IV, endotrophin, which is associated with breast cancer metastatic spread [85,167,168][85][167][168]. Furthermore adipocyte collagen IV has been shown to play a role in the early stages of tumor growth in breast cancer [169]. Recently, it has been shown that breast cancer secreted PAI-1 can stimulate CAA collagen biogenesis and reorganization via the induction of a lysyl hydroxylase protein, PLOD2, facilitating the migration of breast cancer cells along aligned collagen fibers, in vitro and in vivo, further promoting metastasis [170]. Additionally, tumors growing in the adipose tissue-rich microenvironment can induce morphological and functional changes in ADSCs so that they differentiate into a CAF-like myofibroblastic phenotype. Breast cancer cells can induce the differentiation of ADSCs to CAFs involving a mechanism dependent on TGF-β, and these ASC-derived CAF-like cells can promote breast cancer cell motility and invasion in vitro, and expressing high levels of stromal-derived factor 1 (SDF-1), a chemokine associated with a more aggressive and invasive cancer phenotype [171,172][171][172]. During obesity-induced fibrosis, as adipocytes become encased in the rigid ECM, necrosis ensues and dysfunctional adipocytes stimulate the recruitment of macrophages to the site, histologically forming CLS around the dying adipocytes [173]. The contribution of these activated macrophages to ECM production most likely occurs through their effects on other cell types. They are a major source of TGF-β, PDGF and other chemokines that attract and activate more ECM protein producing fibroblast type cells to the adipose tissue [174,175][174][175]. Finally, fibrosis dynamics are tightly regulated by the metalloproteinase (MMP) protein family, which cleave collagenous fibers, enabling matrix remodeling. There are many MMPs associated with obesity [176], in particular MMP-11 (also known as stromelysin-3/ST-3) has been shown to be overexpressed by adipocytes as a result of stimulation by invading breast cancer cells [177]. MMP-11 is important for collagen VI folding and it is also known to negatively regulate adipogenesis and can dedifferentiate adipocytes so that they can acquire a more fibroblast-like phenotype that benefits the invading tumor cells via involvement in adipose tissue fibrosis and ECM remodeling [45,177][45][177].
3.6. Extracellular Vesicles
In addition to the milieu of adipose tissue secreted factors discussed in the previous sections, there is now evidence highlighting the role that adipose tissue derived extracellular vesicles (EVs) play in guiding and enhancing the metastatic process. EVs are lipid membrane enclosed particles, measuring on the nanometer scale, that can be classified as either minute exosomes (<100 nM) or larger microvesicles (<1000 nM), which facilitate the horizontal transfer of cellular cargo, including nucleic acid, proteins, lipids and metabolites between communicating cells [178,179][178][179]. It is not clear whether obesity alters the content of EVs produced by adipocytes, however in the obese setting larger quantities of adipocyte-derived EVs are secreted compared to lean conditions [180]. While the role of cancer cell-derived EVs in manipulating cells in the tumor microenvironment including adipocytes is well established [181[181][182][183],182,183], very little attention has been given to the bidirectional role EVs play in adipocyte-cancer cell communication, and particularly the influence of adipocyte-derived EVs on tumor cell behavior. Nevertheless, a small number of recent studies demonstrate a link between adipocyte EVs and tumor progression in obesity-driven lung cancer [184], breast cancer [185,186][185][186] and melanoma [180,187][180][187].
One way in which adipocytes can promote tumor progression is through metabolic cooperation, by providing a local supply of fatty acids for the process of fatty acid oxidation (FAO) within tumor cells, an emerging favorable metabolic pathway that enhances tumor invasiveness, proliferation and stem cell properties [180,188,189][180][188][189]. In the later stages of tumor progression once the tumor cells have invaded the adipose tissue, secretion of tumor-derived soluble factors can stimulate adipocyte lipolysis and extracellular release of FFAs into the surrounding microenvironment [39,134,188][39][134][188]. Aside from direct release of FAs from adipocytes, EVs released by adipocytes can also be used as a method to transfer molecules including FA substrates and the protein machinery required for FAO to cancer cells either locally or over larger distances, e.g., through the circulation and other tissues.
Epidemiological studies have identified a link between melanoma aggressiveness and obesity [190,191][190][191]. Aptly, subcutaneous adipocytes are one of the main components of the tumor microenvironment of invading melanomas and indeed melanoma cells have been demonstrated to internalize naïve adipocyte (i.e., not previously exposed to cancer cells)-derived EVs, resulting in amplified FAO and an enhanced migratory and invasive tumor cell phenotype [180]. Lazar et al. demonstrated that melanoma cells cultured with adipocyte secreted EVs had an increased ability to form lung metastases in mice xenograft models, with a concomitant upregulation of tumor cell FAO. As previously mentioned obese-derived adipocytes secrete higher numbers of EVs compared to their lean counterparts, thus when comparing the effect of adipocyte EVs derived from obese versus lean conditions, equal concentrations of EV preparations were applied to melanoma cells. Noticeably, in obese conditions only adipocyte-derived EVs elicit a heightened effect on melanoma migration, in addition to enhancing clonogenicity and metastatic potential. Thus differences in the cargo content of these EVs as opposed to the sheer number of vesicles are most likely responsible for this heightened effect. Recent in-depth labeling experiments indicate that around 30% of the proteins within the adipocyte EVs are sufficiently transferred to melanoma tumor cells, and these include proteins involved in EV transport, the transport and storage of FAs, mitochondrial FAO and oxidative phosphorylation [187]. Efforts to understand how an obese state heightens the effect of adipocyte EVs on FAO-induced functions of melanoma cells have revealed that the level of FAO enzymes in melanoma tumor cells is unaltered following uptake of obese versus lean derived EVs. In contrast adipocyte EV supply of FAs and subsequent trafficking to melanoma cells was increased under obese conditions, resulting in enhanced substrate availability and FAO, and the altered mitochondrial dynamics that is critical to melanoma cell migration and invasion. Thus in the obese setting, it is the increased transfer of substrate (e.g., FA) and not machinery (FAO-related enzymes) that enhance FAO in recipient melanoma cancer cells [187].
In whole adipose tissue, aside from mature adipocytes, EVs are found in the supernatant ASCs. While adipocyte EVs are linked to enhanced tumor invasiveness and metastatic potential via lipid metabolism, current literature suggest ADSC secreted EVs play a role in angiogenesis [192,193,194][192][193][194], immune modulation [195,196,197][195][196][197] and tumor development [198]. When exosomes secreted from the preadipocyte cell line, 3T3-L1, are injected into the mammary fat together with breast cancer cells, primary tumor initiation and growth is enhanced. Of particular interest in this review, ASC derived exosomes have been shown to promote proliferation and migration, at least in part through the modulation of Wnt/β-catenin signaling, a key pathway in tumor stemness, EMT and metastasis [199,200][199][200]. ASCs are abundant in the microenvironment of highly metastatic osteosarcoma. Recently ASC exosomes has been shown to increase proliferation, invasion and migration of osteosarcoma cells in vitro, and growth and metastasis in vivo, via an induction of COLGALT2, a prometastatic gene that subsequently activates downstream EMT targets vimentin and MMP-2 and -9 [201]. Studies regarding adipose tissue secreted exosomes, be it adipocytes or ASCs, are limited to a small number of studies and a small number of tumor types. Further research is required to understand the role, if any, of adipose tissue-derived EVs in other cancer types, which share an intimate relationship with adipose tissue and are driven by obesity.
References
- Steele, C.B.; Thomas, C.C.; Henley, S.J.; Massetti, G.M.; Galuska, D.A.; Agurs-Collins, T.; Puckett, M.; Richardson, L.C. Vital Signs: Trends in Incidence of Cancers Associated with Overweight and Obesity—United States, 2005–2014. MMWR Morb. Mortal. Wkly. Rep. 2017, 66, 1052–1058, doi:10.15585/mmwr.mm6639e1.
- Chan, D.S.M.; Vieira, A.R.; Aune, D.; Bandera, E.V.; Greenwood, D.C.; McTiernan, A.; Rosenblatt, D.N.; Thune, I.; Vieira, R.; Norat, T. Body mass index and survival in women with breast cancer—Systematic literature review and meta-analysis of 82 follow-up studies. Ann. Oncol. 2014, 25, 1901–1914.
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 2018, 4, 374–384.
- Renehan, A.G.; Zwahlen, M.; Egger, M. Adiposity and cancer risk: New mechanistic insights from epidemiology. Nat. Rev. Cancer 2015, 15, 484–498.
- Allott, E.H.; Masko, E.M.; Freedland, S.J. Obesity and prostate cancer: Weighing the evidence. Eur. Urol. 2013, 63, 800–809.
- Huffman, D.M.; Johnson, M.S.; Watts, A.; Elgavish, A.; Eltoum, I.A.; Nagy, T.R. Cancer progression in the transgenic adenocarcinoma of mouse prostate mouse is related to energy balance, body mass, and body composition, but not food intake. Cancer Res. 2007, 67, 417–424, doi:10.1158/0008-5472.CAN-06-1244.
- Ewertz, M.; Jensen, M.B.; Gunnarsdóttir, K.Á.; Højris, I.; Jakobsen, E.H.; Nielsen, D.; Stenbygaard, L.E.; Tange, U.B.; Cold, S. Effect of obesity on prognosis after early-stage breast cancer. J. Clin. Oncol. 2011, 29, 25–31, doi:10.1200/JCO.2010.29.7614.
- Kyrgiou, M.; Kalliala, I.; Markozannes, G.; Gunter, M.J.; Paraskevaidis, E.; Gabra, H.; Martin-Hirsch, P.; Tsilidis, K.K. Adiposity and cancer at major anatomical sites: Umbrella review of the literature. BMJ 2017, 356, 477.
- O’Sullivan, J.; Lysaght, J.; Donohoe, C.L.; Reynolds, J.V. Obesity and gastrointestinal cancer: The interrelationship of adipose and tumour microenvironments. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 699–714.
- Fang, X.; Wei, J.; He, X.; Lian, J.; Han, D.; An, P.; Zhou, T.; Liu, S.; Wang, F.; Min, J. Quantitative association between body mass index and the risk of cancer: A global Meta-analysis of prospective cohort studies. Int. J. Cancer 2018, 143, 1595–1603, doi:10.1002/ijc.31553.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674, doi:10.1016/j.cell.2011.02.013.
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027.
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472, doi:10.1038/nature25432.
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686.
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180.
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530, doi:10.1038/nature16064.
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476, doi:10.1038/nature15748.
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 2018, 18, 669–680, doi:10.1038/s41568-018-0056-x.
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 1–17.
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468, doi:10.1038/s41586-018-0040-3.
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769.
- Fox, R.G.; Lytle, N.K.; Jaquish, D.V.; Park, F.D.; Ito, T.; Bajaj, J.; Koechlein, C.S.; Zimdahl, B.; Yano, M.; Kopp, J.L.; et al. Image-based detection and targeting of therapy resistance in pancreatic adenocarcinoma. Nature 2016, 534, 407–411, doi:10.1038/nature17988.
- Balic, M.; Lin, H.; Young, L.; Hawes, D.; Giuliano, A.; McNamara, G.; Datar, R.H.; Cote, R.J. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin. Cancer Res. 2006, 12, 5615–5621, doi:10.1158/1078-0432.CCR-06-0169.
- Grillet, F.; Bayet, E.; Villeronce, O.; Zappia, L.; Lagerqvist, E.L.; Lunke, S.; Charafe-Jauffret, E.; Pham, K.; Molck, C.; Rolland, N.; et al. Circulating tumour cells from patients with colorectal cancer have cancer stem cell hallmarks in ex vivo culture. Gut 2017, 66, 1802–1810, doi:10.1136/gutjnl-2016-311447.
- Aktas, B.; Tewes, M.; Fehm, T.; Hauch, S.; Kimmig, R.; Kasimir-Bauer, S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009, 11, doi:10.1186/bcr2333.
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bäuerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544, doi:10.1038/nbt.2576.
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323, doi:10.1016/j.stem.2007.06.002.
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018, 2018, doi:10.1155/2018/5416923.
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317.
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873, doi:10.1016/S0002-9440(10)65628-3.
- Bockhorn, M.; Jain, R.K.; Munn, L.L. Active versus passive mechanisms in metastasis: Do cancer cells crawl into vessels, or are they pushed? Lancet Oncol. 2007, 8, 444–448.
- Hillen, F.; Griffioen, A.W. Tumour vascularization: Sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007, 26, 489–502, doi:10.1007/s10555-007-9094-7.
- Park, S.Y.; Nam, J.S. The force awakens: Metastatic dormant cancer cells. Exp. Mol. Med. 2020, 52, 569–581.
- Giancotti, F.G. XMechanisms governing metastatic dormancy and reactivation. Cell 2013, 155, 750.
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, doi:10.1126/science.aao4227.
- Choi, J.; Cha, Y.J.; Koo, J.S. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog. Lipid Res. 2018, 69, 11–20.
- Gerber, S.A.; Rybalko, V.Y.; Bigelow, C.E.; Lugade, A.A.; Foster, T.H.; Frelinger, J.G.; Lord, E.M. Preferential attachment of peritoneal tumor metastases to omental immune aggregates and possible role of a unique vascular microenvironment in metastatic survival and growth. Am. J. Pathol. 2006, 169, 1739–1752, doi:10.2353/ajpath.2006.051222.
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503, doi:10.1038/nm.2492.
- Cao, H.; Sekiya, M.; Ertunc, M.E.; Burak, M.F.; Mayers, J.R.; White, A.; Inouye, K.; Rickey, L.M.; Ercal, B.C.; Furuhashi, M.; et al. Adipocyte lipid chaperone aP2 Is a secreted adipokine regulating hepatic glucose production. Cell Metab. 2013, 17, 768–778, doi:10.1016/j.cmet.2013.04.012.
- Hao, J.; Zhang, Y.; Yan, X.; Yan, F.; Sun, Y.; Zeng, J.; Waigel, S.; Yin, Y.; Fraig, M.M.; Egilmez, N.K.; et al. Circulating Adipose Fatty Acid Binding Protein Is a New Link Underlying Obesity-Associated Breast/Mammary Tumor Development. Cell Metab. 2018, 28, 689–705.e5, doi:10.1016/j.cmet.2018.07.006.
- Yan, F.; Shen, N.; Pang, J.X.; Zhang, Y.W.; Rao, E.Y.; Bode, A.M.; Al-Kali, A.; Zhang, D.E.; Litzow, M.R.; Li, B.; et al. Fatty acid-binding protein FABP4 mechanistically links obesity with aggressive AML by enhancing aberrant DNA methylation in AML cells. Leukemia 2017, 31, 1434–1442, doi:10.1038/leu.2016.349.
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 1–15.
- Guerrero, J.; Tobar, N.; Cáceres, M.; Espinoza, L.; Escobar, P.; Dotor, J.; Smith, P.C.; Martínez, J. Soluble factors derived from tumor mammary cell lines induce a stromal mammary adipose reversion in human and mice adipose cells. Possible role of TGF-β1 and TNF-α. Breast Cancer Res. Treat. 2010, 119, 497–508, doi:10.1007/s10549-009-0491-1.
- Andarawewa, K.L.; Motrescu, E.R.; Chenard, M.P.; Gansmuller, A.; Stoll, I.; Tomasetto, C.; Rio, M.C. Stromelysin-3 is a potent negative regulator of adipogenesis participating to cancer cell-adipocyte interaction/crosstalk at the tumor invasive front. Cancer Res. 2005, 65, 10862–10871, doi:10.1158/0008-5472.CAN-05-1231.
- Meunier, P.; Aaron, J.; Edouard, C.; Vignon, G. Osteoporosis and the replacement of cell populations of the marrow by adipose tissue. A quantitative study of 84 iliac bone biopsies. Clin. Orthop. Relat. Res. 1971, 80, 147–154, doi:10.1097/00003086-197110000-00021.
- Scheller, E.L.; Rosen, C.J. What’s the matter with MAT? Marrow adipose tissue, metabolism, and skeletal health. Ann. N. Y. Acad. Sci. 2014, 1311, 14–30, doi:10.1111/nyas.12327.
- Morris, E.V.; Edwards, C.M. The role of bone marrow adipocytes in bone metastasis. J. Bone Oncol. 2016, 5, 121–123, doi:10.1016/j.jbo.2016.03.006.
- Olechnowicz, S.W.Z.; Edwards, C.M. Contributions of the host microenvironment to cancer-induced bone disease. Cancer Res. 2014, 74, 1625–1631.
- Woolthuis, C.M.; Stranahan, A.W.; Park, C.Y.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Jordan, C.T. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37, doi:10.1016/j.stem.2016.06.001.
- Herroon, M.K.; Rajagurubandara, E.; Hardaway, A.L.; Powell, K.; Turchick, A.; Feldmann, D.; Podgorski, I. Bone marrow adipocytes promote tumor growth in bone via FABP4-dependent mechanisms. Oncotarget 2013, 4, 2108–2123, doi:10.18632/oncotarget.1482.
- Diedrich, J.D.; Rajagurubandara, E.; Herroon, M.K.; Mahapatra, G.; Hüttemann, M.; Podgorski, I. Bone marrow adipocytes promote the warburg phenotype in metastatic prostate tumors via HIF-1α activation. Oncotarget 2016, 7, 64854–64877, doi:10.18632/oncotarget.11712.
- Cawthorn, W.P.; Scheller, E.L.; Learman, B.S.; Parlee, S.D.; Simon, B.R.; Mori, H.; Ning, X.; Bree, A.J.; Schell, B.; Broome, D.T.; et al. Bone marrow adipose tissue is an endocrine organ that contributes to increased circulating adiponectin during caloric restriction. Cell Metab. 2014, 20, 368–375, doi:10.1016/j.cmet.2014.06.003.
- Katira, A.; Tan, P.H. Evolving role of adiponectin in cancer-controversies and update. Cancer Biol. Med. 2016, 13, 101–119.
- Batista, M.L.; Olivan, M.; Alcantara, P.S.M.; Sandoval, R.; Peres, S.B.; Neves, R.X.; Silverio, R.; Maximiano, L.F.; Otoch, J.P.; Seelaender, M. Adipose tissue-derived factors as potential biomarkers in cachectic cancer patients. Cytokine 2013, 61, 532–539, doi:10.1016/j.cyto.2012.10.023.
- Tan, P.H.; Tyrrell, H.E.J.; Gao, L.; Xu, D.; Quan, J.; Gill, D.; Rai, L.; Ding, Y.; Plant, G.; Chen, Y.; et al. Adiponectin receptor signaling on dendritic cells blunts antitumor immunity. Cancer Res. 2014, 74, 5711–5722, doi:10.1158/0008-5472.CAN-13-1397.
- Trayhurn, P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol. Rev. 2013, 93, 1–21, doi:10.1152/physrev.00017.2012.
- Chen, B.; Lam, K.S.L.; Wang, Y.; Wu, D.; Lam, M.C.; Shen, J.; Wong, L.; Hoo, R.L.C.; Zhang, J.; Xu, A. Hypoxia dysregulates the production of adiponectin and plasminogen activator inhibitor-1 independent of reactive oxygen species in adipocytes. Biochem. Biophys. Res. Commun. 2006, 341, 549–556, doi:10.1016/j.bbrc.2006.01.004.
- Wang, B.; Wood, I.S.; Trayhurn, P. Hypoxia induces leptin gene expression and secretion in human preadipocytes: Differential effects of hypoxia on adipokine expression by preadipocytes. J. Endocrinol. 2008, 198, 127–134, doi:10.1677/JOE-08-0156.
- Chen, D.C.; Chung, Y.F.; Yeh, Y.T.; Chaung, H.C.; Kuo, F.C.; Fu, O.Y.; Chen, H.Y.; Hou, M.F.; Yuan, S.S.F. Serum adiponectin and leptin levels in Taiwanese breast cancer patients. Cancer Lett. 2006, 237, 109–114, doi:10.1016/j.canlet.2005.05.047.
- Sultana, R.; Kataki, A.C.; Borthakur, B.B.; Basumatary, T.K.; Bose, S. Imbalance in leptin-adiponectin levels and leptin receptor expression as chief contributors to triple negative breast cancer progression in Northeast India. Gene 2017, 621, 51–58, doi:10.1016/j.gene.2017.04.021.
- Barone, I.; Giordano, C.; Bonofiglio, D.; Andò, S.; Catalano, S. The weight of obesity in breast cancer progression and metastasis: Clinical and molecular perspectives. Semin. Cancer Biol. 2020, 60, 274–284.
- Alshaker, H.; Krell, J.; Frampton, A.E.; Waxman, J.; Blyuss, O.; Zaikin, A.; Winkler, M.; Stebbing, J.; Yagüe, E.; Pchejetski, D. Leptin induces upregulation of sphingosine kinase 1 in oestrogen receptor-negative breast cancer via Src family kinase-mediated, janus kinase 2-independent pathway. Breast Cancer Res. 2014, 16, 426, doi:10.1186/s13058-014-0426-6.
- Sabol, R.A.; Bowles, A.C.; Côté, A.; Wise, R.; O’Donnell, B.; Matossian, M.D.; Hossain, F.M.; Burks, H.E.; Del Valle, L.; Miele, L.; et al. Leptin produced by obesity-altered adipose stem cells promotes metastasis but not tumorigenesis of triple-negative breast cancer in orthotopic xenograft and patient-derived xenograft models. Breast Cancer Res. 2019, 21, doi:10.1186/s13058-019-1153-9.
- Chang, C.C.; Wu, M.J.; Yang, J.Y.; Camarillo, I.G.; Chang, C.J. Leptin-STAT3-G9a signaling promotes obesity-mediated breast cancer progression. Cancer Res. 2015, 75, 2375–2386, doi:10.1158/0008-5472.CAN-14-3076.
- Brandon, J.A.; Kraemer, M.; Vandra, J.; Halder, S.; Ubele, M.; Morris, A.J.; Smyth, S.S. Adipose-derived autotaxin regulates inflammation and steatosis associated with diet-induced obesity. PLoS ONE 2019, 14, doi:10.1371/journal.pone.0208099.
- Benesch, M.G.K.; Tang, X.; Brindley, D.N. Autotaxin and breast cancer: Towards overcoming treatment barriers and sequelae. Cancers 2020, 12, 374.
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of Selective Inhibitors of Cancer Stem Cells by High-Throughput Screening. Cell 2009, 138, 645–659, doi:10.1016/j.cell.2009.06.034.
- Shim, S.J.; Shin, E.; Lee, C.-S.; Koo, J.S. The expressions of autotaxin-lysophosphatidate signaling-related proteins in metastatic breast cancer. Int. J. Clin. Exp. Pathol. 2019, 12, 2920–2930.
- Dusaulcy, R.; Rancoule, C.; Grès, S.; Wanecq, E.; Colom, A.; Guigné, C.; Van Meeteren, L.A.; Moolenaar, W.H.; Valet, P.; Saulnier-Blache, J.S. Adipose-specific disruption of autotaxin enhances nutritional fattening and reduces plasma lysophosphatidic acid. J. Lipid Res. 2011, 52, 1247–1255, doi:10.1194/jlr.M014985.
- Rancoule, C.; Dusaulcy, R.; Tréguer, K.; Grès, S.; Guigné, C.; Quilliot, D.; Valet, P.; Saulnier-Blache, J.S. Depot-specific regulation of autotaxin with obesity in human adipose tissue. J. Physiol. Biochem. 2012, 68, 635–644, doi:10.1007/s13105-012-0181-z.
- Benesch, M.G.K.; Macintyre, I.T.K.; McMullen, T.P.W.; Brindley, D.N. Coming of age for autotaxin and lysophosphatidate signaling: Clinical applications for preventing, detecting and targeting tumor-promoting inflammation. Cancers 2018, 10, 73.
- Reeves, V.L.; Trybula, J.S.; Wills, R.C.; Goodpaster, B.H.; Dubé, J.J.; Kienesberger, P.C.; Kershaw, E.E. Serum Autotaxin/ENPP2 correlates with insulin resistance in older humans with obesity. Obesity 2015, 23, 2371–2376, doi:10.1002/oby.21232.
- Azare, J.; Doane, A.; Leslie, K.; Chang, Q.; Berishaj, M.; Nnoli, J.; Mark, K.; Al-Ahmadie, H.; Gerald, W.; Hassimi, M.; et al. Stat3 mediates expression of autotaxin in breast cancer. PLoS ONE 2011, 6, doi:10.1371/journal.pone.0027851.
- Du, J.; Sun, C.; Hu, Z.; Yang, Y.; Zhu, Y.; Zheng, D.; Gu, L.; Lu, X. Lysophosphatidic Acid Induces MDA-MB-231 Breast Cancer Cells Migration through Activation of PI3K/PAK1/ERK Signaling. PLoS ONE 2010, 5, e15940, doi:10.1371/journal.pone.0015940.
- Wu, T.; Van der Kooi, C.; Shah, P.; Charnigo, R.; Huang, C.; Smyth, S.S.; Morris, A.J. Integrin-mediated cell surface recruitment of autotaxin promotes persistent directional cell migration. FASEB J. 2014, 28, 861–870, doi:10.1096/fj.13-232868.
- Cha, Y.J.; Koo, J.S. Adipokines as therapeutic targets in breast cancer treatment. Expert Opin. Ther. Targets 2018, 22, 941–953.
- Benesch, M.G.K.; Tang, X.; Maeda, T.; Ohhata, A.; Zhao, Y.Y.; Kok, B.P.C.; Dewald, J.; Hitt, M.; Curtis, J.M.; McMullen, T.P.W.; et al. Inhibition of autotaxin delays breast tumor growth and lung metastasis in mice. FASEB J. 2014, 28, 2655–2666, doi:10.1096/fj.13-248641.
- Boucharaba, A.; Serre, C.-M.; Grès, S.; Saulnier-Blache, J.S.; Bordet, J.-C.; Guglielmi, J.; Clézardin, P.; Peyruchaud, O. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J. Clin. Invest. 2004, 114, 1714–1725, doi:10.1172/jci22123.
- Zhang, H.; Xu, X.; Gajewiak, J.; Tsukahara, R.; Fujiwara, Y.; Liu, J.; Fells, J.I.; Perygin, D.; Parrill, A.L.; Tigyi, G.; et al. Dual activity lysophosphatidic acid receptor pan-antagonist/autotaxin inhibitor reduces breast cancer cell migration in vitro and causes tumor regression in vivo. Cancer Res. 2009, 69, 5441–5449, doi:10.1158/0008-5472.CAN-09-0302.
- Magkrioti, C.; Galaris, A.; Kanellopoulou, P.; Stylianaki, E.A.; Kaffe, E.; Aidinis, V. Autotaxin and chronic inflammatory diseases. J. Autoimmun. 2019, 104, 102327.
- Maher, T.M.; Kreuter, M.; Lederer, D.J.; Brown, K.K.; Wuyts, W.; Verbruggen, N.; Stutvoet, S.; Fieuw, A.; Ford, P.; Abi-Saab, W.; et al. Rationale, design and objectives of two phase III, randomised, placebo-controlled studies of GLPG1690, a novel autotaxin inhibitor, in idiopathic pulmonary fibrosis (ISABELA 1 and 2). BMJ Open Respir. Res. 2019, 6, doi:10.1136/bmjresp-2019-000422.
- Maher, T.M.; van der Aar, E.M.; Van de Steen, O.; Allamassey, L.; Desrivot, J.; Dupont, S.; Fagard, L.; Ford, P.; Fieuw, A.; Wuyts, W. Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): A phase 2a randomised placebo-controlled trial. Lancet Respir. Med. 2018, 6, 627–635, doi:10.1016/S2213-2600(18)30181-4.
- Tang, X.; Wuest, M.; Benesch, M.G.K.; Dufour, J.; Zhao, Y.Y.; Curtis, J.M.; Monjardet, A.; Heckmann, B.; Murray, D.; Wuest, F.; et al. Inhibition of autotaxin with GLPG1690 increases the efficacy of radiotherapy and chemotherapy in a mouse model of breast cancer. Mol. Cancer Ther. 2020, 19, 63–74, doi:10.1158/1535-7163.MCT-19-0386.
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465, doi:10.1158/0008-5472.CAN-10-3323.
- Kern, L.; Mittenbühler, M.J.; Vesting, A.J.; Ostermann, A.L.; Wunderlich, C.M.; Wunderlich, F.T. Obesity-induced TNFα and IL-6 signaling: The missing link between obesity and inflammation-driven liver and colorectal cancers. Cancers 2019, 11, 24.
- Arendt, L.M.; McCready, J.; Keller, P.J.; Baker, D.D.; Naber, S.P.; Seewaldt, V.; Kuperwasser, C. Obesity promotes breast cancer by CCL2-mediated macrophage recruitment and angiogenesis. Cancer Res. 2013, 73, 6080–6093, doi:10.1158/0008-5472.CAN-13-0926.
- Tsuyada, A.; Chow, A.; Wu, J.; Somlo, G.; Chu, P.; Loera, S.; Luu, T.; Li, A.X.; Wu, X.; Ye, W.; et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012, 72, 2768–2779, doi:10.1158/0008-5472.CAN-11-3567.
- D’Esposito, V.D.; Liguoro, D.; Ambrosio, M.R.; Collina, F.; Cantile, M.; Spinelli, R.; Raciti, G.A.; Miele, C.; Valentino, R.; Campiglia, P.; et al. Adipose microenvironment promotes triple negative breast cancer cell invasiveness and dissemination by producing CCL5. Oncotarget 2016, 7, 24495–24509, doi:10.18632/oncotarget.8336.
- Velasco-Velázquez, M.; Jiao, X.; De La Fuente, M.; Pestell, T.G.; Ertel, A.; Lisanti, M.P.; Pestell, R.G. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 2012, 72, 3839–3850, doi:10.1158/0008-5472.CAN-11-3917.
- Rybinska, I.; Agresti, R.; Trapani, A.; Tagliabue, E.; Triulzi, T. Adipocytes in Breast Cancer, the Thick and the Thin. Cells 2020, 9, 560, doi:10.3390/cells9030560.
- Bastard, J.-P.; Jardel, C.; Bruckert, E.; Blondy, P.; Capeau, J.; Laville, M.; Vidal, H.; Hainque, B. Elevated Levels of Interleukin 6 Are Reduced in Serum and Subcutaneous Adipose Tissue of Obese Women after Weight Loss*. J. Clin. Endocrinol. Metab. 2000, 85, 3338–3342, doi:10.1210/jcem.85.9.6839.
- Kim, H.S.; Jung, M.; Choi, S.K.; Woo, J.; Piao, Y.J.; Hwang, E.H.; Kim, H.; Kim, S.J.; Moon, W.K. IL-6-mediated cross-talk between human preadipocytes and ductal carcinoma in situ in breast cancer progression. J. Exp. Clin. Cancer Res. 2018, 37, 200, doi:10.1186/s13046-018-0867-3.
- Sansone, P.; Storci, G.; Tavolari, S.; Guarnieri, T.; Giovannini, C.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Paterini, P.; Marcu, K.B.; et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Invest. 2007, 117, 3988–4002, doi:10.1172/JCI32533.
- Marotta, L.L.C.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44 +CD24- stem cell-like breast cancer cells in human tumors. J. Clin. Invest. 2011, 121, 2723–2735, doi:10.1172/JCI44745.
- Bergmann, J.; Müller, M.; Baumann, N.; Reichert, M.; Heneweer, C.; Bolik, J.; Lücke, K.; Gruber, S.; Carambia, A.; Boretius, S.; et al. IL-6 trans-signaling is essential for the development of hepatocellular carcinoma in mice. Hepatology 2017, 65, 89–103, doi:10.1002/hep.28874.
- Wang, C.-Q.; Sun, H.-T.; Gao, X.-M.; Ren, N.; Sheng, Y.-Y.; Wang, Z.; Zheng, Y.; Wei, J.-W.; Zhang, K.-L.; Yu, X.-X.; et al. Interleukin-6 enhances cancer stemness and promotes metastasis of hepatocellular carcinoma via up-regulating osteopontin expression. Am. J. Cancer Res. 2016, 6, 1873–1889.
- Lukens, J.R.; Dixit, V.D.; Kanneganti, T.D. Inflammasome activation in obesity-related inflammatory diseases and autoimmunity. Discov. Med. 2011, 12, 65–74.
- Coleman, R.E.; Marshall, H.; Cameron, D.; Dodwell, D.; Burkinshaw, R.; Keane, M.; Gil, M.; Houston, S.J.; Grieve, R.J.; Barrett-Lee, P.J.; et al. Breast-cancer adjuvant therapy with zoledronic acid. N. Engl. J. Med. 2011, 365, 1396–1405, doi:10.1056/NEJMoa1105195.
- Tulotta, C.; Ottewell, P. The role of IL-1B in breast cancer bone metastasis. Endocr. Relat. Cancer 2018, 25, R421–R434.
- Nutter, F.; Holen, I.; Brown, H.K.; Cross, S.S.; Evans, C.A.; Walker, M.; Coleman, R.E.; Westbrook, J.A.; Selby, P.J.; Brown, J.E.; et al. Different molecular profiles are associated with breast cancer cell homing compared with colonisation of bone: Evidence using a novel bone-seeking cell line. Endocr. Relat. Cancer 2014, 21, 327–341, doi:10.1530/ERC-13-0158.
- Hagemann, T.; Wilson, J.; Kulbe, H.; Li, N.F.; Leinster, D.A.; Charles, K.; Klemm, F.; Pukrop, T.; Binder, C.; Balkwill, F.R. Macrophages Induce Invasiveness of Epithelial Cancer Cells Via NF-κB and JNK. J. Immunol. 2005, 175, 1197–1205, doi:10.4049/jimmunol.175.2.1197.
- Chua, H.L.; Bhat-Nakshatri, P.; Clare, S.E.; Morimiya, A.; Badve, S.; Nakshatri, H. NF-κB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: Potential involvement of ZEB-1 and ZEB-2. Oncogene 2007, 26, 711–724, doi:10.1038/sj.onc.1209808.
- Sturm, J.W.; Magdeburg, R.; Berger, K.; Petruch, B.; Samel, S.; Bönninghoff, R.; Keese, M.; Hafner, M.; Post, S. Influence of TNFA on the formation of liver metastases in a syngenic mouse model. Int. J. Cancer 2003, 107, 11–21, doi:10.1002/ijc.11320.
- Ham, B.; Fernandez, M.C.; D’Costa, Z.; Brodt, P. The diverse roles of the TNF axis in cancer progression and metastasis. Trends Cancer Res. 2016, 11, 1–27.
- Faria, S.S.; Corrêa, L.H.; Heyn, G.S.; de Sant’Ana, L.P.; Almeida, R.D.N.; Magalhães, K.G. Obesity and breast cancer: The role of crown-like structures in breast adipose tissue in tumor progression, prognosis, and therapy. J. Breast Cancer 2020, 23, 233–245.
- Berstein, L.M.; Iyevleva, A.G.; Mukhina, M.S.; Vasilyev, D.A.; Poroshina, T.E. Features of omental adipose tissue in endometrial cancer patients with ‘standard’ or ‘metabolically healthy’ obesity: Associations with tumor process characteristics. Springerplus 2016, 5, 1900, doi:10.1186/s40064-016-3582-6.
- Gucalp, A.; Iyengar, N.M.; Zhou, X.K.; Giri, D.D.; Falcone, D.J.; Wang, H.; Williams, S.; Krasne, M.D.; Yaghnam, I.; Kunzel, B.; et al. Periprostatic adipose inflammation is associated with high-grade prostate cancer. Prostate Cancer Prostatic Dis. 2017, 20, 418–423, doi:10.1038/pcan.2017.31.
- Itoh, M.; Kato, H.; Suganami, T.; Konuma, K.; Marumoto, Y.; Terai, S.; Sakugawa, H.; Kanai, S.; Hamaguchi, M.; Fukaishi, T.; et al. Hepatic crown-like structure: A unique histological feature in non-alcoholic steatohepatitis in mice and humans. PLoS ONE 2013, 8, doi:10.1371/journal.pone.0082163.
- Mullooly, M.; Yang, H.P.; Falk, R.T.; Nyante, S.J.; Cora, R.; Pfeiffer, R.M.; Radisky, D.C.; Visscher, D.W.; Hartmann, L.C.; Carter, J.M.; et al. Relationship between crown-like structures and sex-steroid hormones in breast adipose tissue and serum among postmenopausal breast cancer patients. Breast Cancer Res. 2017, 19, doi:10.1186/s13058-016-0791-4.
- Cortellini, A.; Bersanelli, M.; Buti, S.; Cannita, K.; Santini, D.; Perrone, F.; Giusti, R.; Tiseo, M.; Michiara, M.; Di Marino, P.; et al. A multicenter study of body mass index in cancer patients treated with anti-PD-1/PD-L1 immune checkpoint inhibitors: When overweight becomes favorable. J. Immunother. Cancer 2019, 7, 57, doi:10.1186/s40425-019-0527-y.
- Plaks, V.; Kong, N.; Werb, Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell 2015, 16, 225–238, doi:10.1016/j.stem.2015.02.015.
- Zhang, S.; Yang, X.; Wang, L.; Zhang, C. Interplay between inflammatory tumor microenvironment and cancer stem cells (Review). Oncol. Lett. 2018, 16, 679–686.
- Lee, S.H.; Hong, H.S.; Liu, Z.X.; Kim, R.H.; Kang, M.K.; Park, N.-H.; Shin, K.-H. TNFα enhances cancer stem cell-like phenotype via Notch-Hes1 activation in oral squamous cell carcinoma cells. Biochem. Biophys. Res. Commun. 2012, 424, 58–64, doi:10.1016/j.bbrc.2012.06.065.
- Ostyn, P.; El Machhour, R.; Begard, S.; Kotecki, N.; Vandomme, J.; Flamenco, P.; Segard, P.; Masselot, B.; Formstecher, P.; Touil, Y.; et al. Transient TNF regulates the self-renewing capacity of stem-like label-retaining cells in sphere and skin equivalent models of melanoma. Cell Commun. Signal. 2014, 12, doi:10.1186/s12964-014-0052-z.
- Zhao, X.; Ma, L.; Dai, L.; Zuo, D.; Li, X.; Zhu, H.; Xu, F. TNF-α promotes the malignant transformation of intestinal stem cells through the NF-κB and Wnt/β-catenin signaling pathways. Oncol. Rep. 2020, 44, 577–588, doi:10.3892/or.2020.7631.
- Storci, G.; Sansone, P.; Mari, S.; D’Uva, G.; Tavolari, S.; Guarnieri, T.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Chieco, P.; et al. TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J. Cell. Physiol. 2010, 225, 682–691, doi:10.1002/jcp.22264.
- Liu, W.; Lu, X.; Shi, P.; Yang, G.; Zhou, Z.; Li, W.; Mao, X.; Jiang, D.; Chen, C. TNF-α increases breast cancer stem-like cells through up-regulating TAZ expression via the non-canonical NF-κB pathway. Sci. Rep. 2020, 10, 1–11, doi:10.1038/s41598-020-58642-y.
- Teslow, E.A.; Mitrea, C.; Bao, B.; Mohammad, R.M.; Polin, L.A.; Dyson, G.; Purrington, K.S.; Bollig‐Fischer, A. Obesity‐induced MBD2_v2 expression promotes tumor‐initiating triple‐negative breast cancer stem cells. Mol. Oncol. 2019, 13, 894–908, doi:10.1002/1878-0261.12444@10.1002/(ISSN)1878-0261.WORLD-CANCER-DAY-2020.
- Fukumura, D.; Ushiyama, A.; Duda, D.G.; Xu, L.; Tam, J.; Krishna, V.; Chatterjee, K.; Garkavtsev, I.; Jain, R.K. Paracrine regulation of angiogenesis and adipocyte differentiation during in vivo adipogenesis. Circ. Res. 2003, 93, doi:10.1161/01.res.0000099243.20096.fa.
- Fukumura, D.; Incio, J.; Shankaraiah, R.C.; Jain, R.K. Obesity and Cancer: An Angiogenic and Inflammatory Link. Microcirculation 2016, 23, 191–206.
- Zhou, W.; Guo, S.; Gonzalez-Perez, R.R. Leptin pro-angiogenic signature in breast cancer is linked to IL-1 signalling. Br. J. Cancer 2011, 104, 128–137, doi:10.1038/sj.bjc.6606013.
- Kolb, R.; Kluz, P.; Tan, Z.W.; Borcherding, N.; Bormann, N.; Vishwakarma, A.; Balcziak, L.; Zhu, P.; Davies, B.S.; Gourronc, F.; et al. Obesity-associated inflammation promotes angiogenesis and breast cancer via angiopoietin-like 4. Oncogene 2019, 38, 2351–2363, doi:10.1038/s41388-018-0592-6.
- Lohmann, A.E.; Chia, S. Patients with metastatic breast cancer using bevacizumab as a treatment: Is there still a role for it? Curr. Treat. Options Oncol. 2012, 13, 249–262, doi:10.1007/s11864-012-0181-9.
- Jain, R.K.; Duda, D.G.; Willett, C.G.; Sahani, D.V.; Zhu, A.X.; Loeffler, J.S.; Batchelor, T.T.; Sorensen, A.G. Biomarkers of response and resistance to antiangiogenic therapy. Nat. Rev. Clin. Oncol. 2009, 6, 327–338.
- Incio, J.; Ligibel, J.A.; McManus, D.T.; Suboj, P.; Jung, K.; Kawaguchi, K.; Pinter, M.; Babykutty, S.; Chin, S.M.; Vardam, T.D.; et al. Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci. Transl. Med. 2018, 10, 945, doi:10.1126/scitranslmed.aag0945.
- Fernández-Cortés, M.; Delgado-Bellido, D.; Oliver, F.J. Vasculogenic Mimicry: Become an Endothelial Cell “But Not So Much.” Front. Oncol. 2019, 9, 803, doi:10.3389/fonc.2019.00803.
- Coelho, P.; Almeida, J.; Prudêncio, C.; Fernandes, R.; Soares, R. Effect of Adipocyte Secretome in Melanoma Progression and Vasculogenic Mimicry. J. Cell. Biochem. 2016, 117, 1697–1706, doi:10.1002/jcb.25463.
- Marín, D.; Sabater, B. The cancer Warburg effect may be a testable example of the minimum entropy production rate principle. Phys. Biol. 2017, 14, 024001, doi:10.1088/1478-3975/aa64a7.
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825.
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001, doi:10.4161/cc.8.23.10238.
- Armignacco, R.; Cantini, G.; Poli, G.; Guasti, D.; Nesi, G.; Romagnoli, P.; Mannell, M.; Luconi, M. The adipose stem cell as a novel metabolic actor in adrenocortical carcinoma progression: Evidence from an in vitro tumor microenvironment crosstalk model. Cancers 2019, 11, 11, doi:10.3390/cancers11121931.
- Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; Lisanti, M.P. Ketone body utilization drives tumor growth and metastasis. Cell Cycle 2012, 11, 3964–3971, doi:10.4161/cc.22137.
- Wang, Y.Y.; Attané, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, doi:10.1172/jci.insight.87489.
- Zaidi, N.; Lupien, L.; Kuemmerle, N.B.; Kinlaw, W.B.; Swinnen, J.V.; Smans, K. Lipogenesis and lipolysis: The pathways exploited by the cancer cells to acquire fatty acids. Prog. Lipid Res. 2013, 52, 585–589.
- Zhang, M.; Di Martino, J.S.; Bowman, R.L.; Campbell, N.R.; Baksh, S.C.; Simon-Vermot, T.; Kim, I.S.; Haldeman, P.; Mondal, C.; Yong-Gonzales, V.; et al. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discov. 2018, 8, 1006–1025, doi:10.1158/2159-8290.CD-17-1371.
- Shafat, M.S.; Oellerich, T.; Mohr, S.; Robinson, S.D.; Edwards, D.R.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332, doi:10.1182/blood-2016-08-734798.
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31.
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180.
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab. 2016, 24, 685–700, doi:10.1016/j.cmet.2016.10.011.
- Petrus, P.; Lecoutre, S.; Dollet, L.; Wiel, C.; Sulen, A.; Gao, H.; Tavira, B.; Laurencikiene, J.; Rooyackers, O.; Checa, A.; et al. Glutamine Links Obesity to Inflammation in Human White Adipose Tissue. Cell Metab. 2020, 31, 375–390.e11, doi:10.1016/j.cmet.2019.11.019.
- Recouvreux, M.V.; Moldenhauer, M.R.; Galenkamp, K.M.O.; Jung, M.; James, B.; Zhang, Y.; Lowy, A.; Bagchi, A.; Commisso, C. Glutamine depletion regulates Slug to promote EMT and metastasis in pancreatic cancer. J. Exp. Med. 2020, 217, doi:10.1084/jem.20200388.
- Pavlova, N.N.; Hui, S.; Ghergurovich, J.M.; Fan, J.; Intlekofer, A.M.; White, R.M.; Rabinowitz, J.D.; Thompson, C.B.; Zhang, J. As Extracellular Glutamine Levels Decline, Asparagine Becomes an Essential Amino Acid. Cell Metab. 2018, 27, 428–438.e5, doi:10.1016/j.cmet.2017.12.006.
- Luo, M.; Brooks, M.; Wicha, M.S. Asparagine and Glutamine: Co-conspirators Fueling Metastasis. Cell Metab. 2018, 27, 947–949.
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381, doi:10.1038/nature25465.
- Rizi, B.S.; Caneba, C.; Nowicka, A.; Nabiyar, A.W.; Liu, X.; Chen, K.; Klopp, A.; Nagrath, D. Nitric oxide mediates metabolic coupling of omentum-derived adipose stroma to ovarian and endometrial cancer cells. Cancer Res. 2015, 75, 456–471, doi:10.1158/0008-5472.CAN-14-1337.
- Al-Koussa, H.; El Mais, N.; Maalouf, H.; Abi-Habib, R.; El-Sibai, M. Arginine deprivation: A potential therapeutic for cancer cell metastasis? A review. Cancer Cell Int. 2020, 20, 150.
- You, J.; Chen, W.; Chen, J.; Zheng, Q.; Dong, J.; Zhu, Y. The oncogenic role of ARG1 in progression and metastasis of hepatocellular carcinoma. BioMed Res. Int. 2018, 2018, doi:10.1155/2018/2109865.
- Zaytouni, T.; Tsai, P.Y.; Hitchcock, D.S.; Dubois, C.D.; Freinkman, E.; Lin, L.; Morales-Oyarvide, V.; Lenehan, P.J.; Wolpin, B.M.; Mino-Kenudson, M.; et al. Critical role for arginase 2 in obesity-Associated pancreatic cancer. Nat. Commun. 2017, 8, 1–12, doi:10.1038/s41467-017-00331-y.
- Spinelli, J.B.; Yoon, H.; Ringel, A.E.; Jeanfavre, S.; Clish, C.B.; Haigis, M.C. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 2017, 358, 941–946, doi:10.1126/science.aam9305.
- Thomas, D.D.; Corkey, B.E.; Istfan, N.W.; Apovian, C.M. Hyperinsulinemia: An early indicator of metabolic dysfunction. J. Endocr. Soc. 2019, 3, 1727–1747.
- Ireland, L.; Santos, A.; Campbell, F.; Figueiredo, C.; Hammond, D.; Ellies, L.G.; Weyer-Czernilofsky, U.; Bogenrieder, T.; Schmid, M.; Mielgo, A. Blockade of insulin-like growth factors increases efficacy of paclitaxel in metastatic breast cancer. Oncogene 2018, 37, 2022–2036, doi:10.1038/s41388-017-0115-x.
- Malaguarnera, R.; Belfiore, A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front. Endocrinol. 2014, 5, 10.
- Baserga, R. The decline and fall of the IGF-I receptor. J. Cell. Physiol. 2013, 228, 675–679, doi:10.1002/jcp.24217.
- Tong, Y.; Wu, J.; Huang, O.; He, J.; Zhu, L.; Chen, W.; Li, Y.; Chen, X.; Shen, K. IGF-1 Interacted With Obesity in Prognosis Prediction in HER2-Positive Breast Cancer Patients. Front. Oncol. 2020, 10, doi:10.3389/fonc.2020.00550.
- Wu, Y.; Brodt, P.; Sun, H.; Mejia, W.; Novosyadlyy, R.; Nunez, N.; Chen, X.; Mendoza, A.; Hong, S.H.; Khanna, C.; et al. Insulin-like growth factor-I regulates the liver microenvironment in obese mice and promotes liver metastasis. Cancer Res. 2010, 70, 57–67, doi:10.1158/0008-5472.CAN-09-2472.
- Rayes, R.F.; Milette, S.; Fernandez, M.C.; Ham, B.; Wang, N.; Bourdeau, F.; Perrino, S.; Yakar, S.; Brodt, P. Loss of neutrophil polarization in colon carcinoma liver metastases of mice with an inducible, liver-specific IGF-I deficiency. Oncotarget 2018, 9, 15691–15704, doi:10.18632/oncotarget.24593.
- Ruiz-Ojeda, F.J.; Méndez-Gutiérrez, A.; Aguilera, C.M.; Plaza-Díaz, J. Extracellular matrix remodeling of adipose tissue in obesity and metabolic diseases. Int. J. Mol. Sci. 2019, 20, 4888.
- Buechler, C.; Krautbauer, S.; Eisinger, K. Adipose tissue fibrosis. World J. Diabetes 2015, 6, 548, doi:10.4239/wjd.v6.i4.548.
- Hasegawa, Y.; Ikeda, K.; Chen, Y.; Alba, D.L.; Stifler, D.; Shinoda, K.; Hosono, T.; Maretich, P.; Yang, Y.; Ishigaki, Y.; et al. Repression of Adipose Tissue Fibrosis through a PRDM16-GTF2IRD1 Complex Improves Systemic Glucose Homeostasis. Cell Metab. 2018, 27, 180–194.e6, doi:10.1016/j.cmet.2017.12.005.
- Oikonomou, E.K.; Antoniades, C. The role of adipose tissue in cardiovascular health and disease. Nat. Rev. Cardiol. 2019, 16, 83–99.
- Moorman, A.M.; Vink, R.; Heijmans, H.J.; Van Der Palen, J.; Kouwenhoven, E.A. The prognostic value of tumour-stroma ratio in triple-negative breast cancer. Eur. J. Surg. Oncol. 2012, 38, 307–313, doi:10.1016/j.ejso.2012.01.002.
- Zippi, M.; De Toma, G.; Minervini, G.; Cassieri, C.; Pica, R.; Colarusso, D.; Stock, S.; Crispino, P. Desmoplasia influenced recurrence of disease and mortality in stage III colorectal cancer within five years after surgery and adjuvant therapy. Saudi J. Gastroenterol. 2017, 23, 39, doi:10.4103/1319-3767.199114.
- Incio, J.; Liu, H.; Suboj, P.; Chin, S.M.; Chen, I.X.; Pinter, M.; Ng, M.R.; Nia, H.T.; Grahovac, J.; Kao, S.; et al. Obesity-induced inflammation and desmoplasia promote pancreatic cancer progression and resistance to chemotherapy. Cancer Discov. 2016, 6, 852–869, doi:10.1158/2159-8290.CD-15-1177.
- Verset, L.; Tommelein, J.; Lopez, X.M.; Decaestecker, C.; Boterberg, T.; De Vlieghere, E.; Salmon, I.; Mareel, M.; Bracke, M.; De Wever, O.; et al. Impact of neoadjuvant therapy on cancer-associated fibroblasts in rectal cancer. Radiother. Oncol. 2015, 116, 449–454, doi:10.1016/j.radonc.2015.05.007.
- Arkan, M.C. Cancer: Fat and the fate of pancreatic tumours. Nature 2016, 536, 157–158.
- Bochet, L.; Lehuédé, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Gonidec, S. Le; et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013, 73, 5657–5668, doi:10.1158/0008-5472.CAN-13-0530.
- Park, J.; Scherer, P.E. Adipocyte-derived endotrophin promotes malignant tumor progression. J. Clin. Invest. 2012, 122, 4243–4256, doi:10.1172/JCI63930.
- Iyengar, P.; Espina, V.; Williams, T.W.; Lin, Y.; Berry, D.; Jelicks, L.A.; Lee, H.; Temple, K.; Graves, R.; Pollard, J.; et al. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J. Clin. Invest. 2005, 115, 1163–1176, doi:10.1172/JCI23424.
- Wei, X.; Li, S.; He, J.; Du, H.; Liu, Y.; Yu, W.; Hu, H.; Han, L.; Wang, C.; Li, H.; et al. Tumor-secreted PAI-1 promotes breast cancer metastasis via the induction of adipocyte-derived collagen remodeling. Cell Commun. Signal. 2019, 17, 58, doi:10.1186/s12964-019-0373-z.
- Jotzu, C.; Alt, E.; Welte, G.; Li, J.; Hennessy, B.T.; Devarajan, E.; Krishnappa, S.; Pinilla, S.; Droll, L.; Song, Y.H. Adipose tissue derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor derived factors. Cell. Oncol. 2011, 34, 55–67, doi:10.1007/s13402-011-0012-1.
- Kang, H.; Watkins, G.; Parr, C.; Douglas-Jones, A.; Mansel, R.E.; Jiang, W.G. Stromal cell derived factor-1: Its influence on invasiveness and migration of breast cancer cells in vitro, and its association with prognosis and survival in human breast cancer. Breast Cancer Res. 2005, 7, doi:10.1186/bcr1022.
- Cozzo, A.J.; Fuller, A.M.; Makowski, L. Contribution of adipose tissue to development of cancer. Compr. Physiol. 2018, 8, 237–282, doi:10.1002/cphy.c170008.
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257.
- Corrêa, L.H.; Corrêa, R.; Farinasso, C.M.; de Sant’Ana, D.L.P.; Magalhães, K.G. Adipocytes and macrophages interplay in the orchestration of tumor microenvironment: New implications in cancer progression. Front. Immunol. 2017, 8, 1.
- Jaoude, J.; Koh, Y. Matrix metalloproteinases in exercise and obesity. Vasc. Health Risk Manag. 2016, 12, 287–295.
- Motrescu, E.R.; Blaise, S.; Etique, N.; Messaddeq, N.; Chenard, M.P.; Stoll, I.; Tomasetto, C.; Rio, M.C. Matrix metalloproteinase-11/stromelysin-3 exhibits collagenolytic function against collagen VI under normal and malignant conditions. Oncogene 2008, 27, 6347–6355, doi:10.1038/onc.2008.218.
- De Lope, L.R.; Alcíbar, O.L.; López, A.A.; Hergueta-Redondo, M.; Peinado, H. Tumour–adipose tissue crosstalk: Fuelling tumour metastasis by extracellular vesicles. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160485.
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289.
- Lazar, I.; Clement, E.; Dauvillier, S.; Milhas, D.; Ducoux-Petit, M.; LeGonidec, S.; Moro, C.; Soldan, V.; Dalle, S.; Balor, S.; et al. Adipocyte Exosomes Promote Melanoma Aggressiveness through Fatty Acid Oxidation: A Novel Mechanism Linking Obesity and Cancer. Cancer Res. 2016, 76, 4051–4057, doi:10.1158/0008-5472.CAN-16-0651.
- Milane, L.; Singh, A.; Mattheolabakis, G.; Suresh, M.; Amiji, M.M. Exosome mediated communication within the tumor microenvironment. J. Control. Release 2015, 219, 278–294.
- Sun, S.; Wu, Q.; Li, J.; Li, Z.; Sun, S.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Zhang, Y.; et al. Exosomes from the tumour-adipocyte interplay stimulate beige/brown differentiation and reprogram metabolism in stromal adipocytes to promote tumour progression. J. Exp. Clin. Cancer Res. 2019, 38, 223, doi:10.1186/s13046-019-1210-3.
- Sagar, G.; Sah, R.P.; Javeed, N.; Dutta, S.K.; Smyrk, T.C.; Lau, J.S.; Giorgadze, N.; Tchkonia, T.; Kirkland, J.L.; Chari, S.T.; et al. Pathogenesis of pancreatic cancer exosome-induced lipolysis in adipose tissue. Gut 2016, 65, 1165–1174, doi:10.1136/gutjnl-2014-308350.
- Wang, J.; Wu, Y.; Guo, J.; Fei, X.; Yu, L.; Ma, S. Adipocyte-derived exosomes promote lung cancer metastasis by increasing MMP9 activity via transferring MMP3 to lung cancer cells. Oncotarget 2017, 8, 81880–81891, doi:10.18632/oncotarget.18737.
- Wang, S.; Su, X.; Xu, M.; Xiao, X.; Li, X.; Li, H.; Keating, A.; Zhao, R.C. Exosomes secreted by mesenchymal stromal/stem cell-derived adipocytes promote breast cancer cell growth via activation of Hippo signaling pathway. Stem Cell Res. Ther. 2019, 10, 117, doi:10.1186/s13287-019-1220-2.
- Benito-Martin, A.; Paik, P.; Mushannen, M.; Bhardwaj, P.; Oshchepkova, S.; Spector, J.; Brown, K.A. SAT-126 Breast Adipose Tissue Extracellular Vesicles from Obese Women Increase Breast Cancer Aggressiveness—A Novel Mechanism for the Obesity-Breast Cancer Link. J. Endocr. Soc. 2020, 4, doi:10.1210/jendso/bvaa046.1831.
- Clement, E.; Lazar, I.; Attané, C.; Carrié, L.; Dauvillier, S.; Ducoux-Petit, M.; Menneteau, T.; Moutahir, M.; Le Gonidec, S.; Dalle, S.; et al. Adipocyte vesicles: ‘all-in-one’ packages that stimulate tumor mitochondrial metabolism and dynamics. bioRxiv 2019, 649327, doi:10.1101/649327.
- Balaban, S.; Shearer, R.F.; Lee, L.S.; van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017, 5, 1, doi:10.1186/s40170-016-0163-7.
- Kuo, C.Y.; Ann, D.K. When fats commit crimes: Fatty acid metabolism, cancer stemness and therapeutic resistance. Cancer Commun. 2018, 38, 47.
- Skowron, F.; Bérard, F.; Balme, B.; Maucort-Boulch, D. Role of obesity on the thickness of primary cutaneous melanoma. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 262–269, doi:10.1111/jdv.12515.
- Clement, E.; Lazar, I.; Muller, C.; Nieto, L. Obesity and melanoma: Could fat be fueling malignancy? Pigment Cell Melanoma Res. 2017, 30, 294–306, doi:10.1111/pcmr.12584.
- Lopatina, T.; Bruno, S.; Tetta, C.; Kalinina, N.; Porta, M.; Camussi, G. Platelet-derived growth factor regulates the secretion of extracellular vesicles by adipose mesenchymal stem cells and enhances their angiogenic potential. Cell Commun. Signal. 2014, 12, 26, doi:10.1186/1478-811X-12-26.
- Ren, S.; Chen, J.; Duscher, D.; Liu, Y.; Guo, G.; Kang, Y.; Xiong, H.; Zhan, P.; Wang, Y.; Wang, C.; et al. Microvesicles from human adipose stem cells promote wound healing by optimizing cellular functions via AKT and ERK signaling pathways. Stem Cell Res. Ther. 2019, 10, 47, doi:10.1186/s13287-019-1152-x.
- An, Y.; Zhao, J.; Nie, F.; Qin, Z.; Xue, H.; Wang, G.; Li, D. Exosomes from Adipose-Derived Stem Cells (ADSCs) Overexpressing miR-21 Promote Vascularization of Endothelial Cells. Sci. Rep. 2019, 9, 1–10, doi:10.1038/s41598-019-49339-y.
- Liu, Z.; Xu, Y.; Wan, Y.; Gao, J.; Chu, Y.; Li, J. Exosomes from adipose-derived mesenchymal stem cells prevent cardiomyocyte apoptosis induced by oxidative stress. Cell Death Discov. 2019, 5, 1–7, doi:10.1038/s41420-019-0159-5.
- Blazquez, R.; Sanchez-Margallo, F.M.; de la Rosa, O.; Dalemans, W.; Álvarez, V.; Tarazona, R.; Casado, J.G. Immunomodulatory potential of human adipose mesenchymal stem cells derived exosomes on in vitro stimulated T cells. Front. Immunol. 2014, 5, 556, doi:10.3389/fimmu.2014.00556.
- Hong, P.; Yang, H.; Wu, Y.; Li, K.; Tang, Z. The functions and clinical application potential of exosomes derived from adipose mesenchymal stem cells: A comprehensive review. Stem Cell Res. Ther. 2019, 10, 1–12.
- Gernapudi, R.; Yao, Y.; Zhang, Y.; Wolfson, B.; Roy, S.; Duru, N.; Eades, G.; Yang, P.; Zhou, Q. Targeting exosomes from preadipocytes inhibits preadipocyte to cancer stem cell signaling in early-stage breast cancer. Breast Cancer Res. Treat. 2015, 150, 685–695, doi:10.1007/s10549-015-3326-2.
- Lin, R.; Wang, S.; Zhao, R.C. Exosomes from human adipose-derived mesenchymal stem cells promote migration through Wnt signaling pathway in a breast cancer cell model. Mol. Cell. Biochem. 2013, 383, 13–20, doi:10.1007/s11010-013-1746-z.
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473.
- Wang, Y.; Chu, Y.; Li, K.; Zhang, G.; Guo, Z.; Wu, X.; Qiu, C.; Li, Y.; Wan, X.; Sui, J.; et al. Exosomes Secreted by Adipose-Derived Mesenchymal Stem Cells Foster Metastasis and Osteosarcoma Proliferation by Increasing COLGALT2 Expression. Front. Cell Dev. Biol. 2020, 8, 353, doi:10.3389/fcell.2020.00353.

