1000/1000
Hot
Most Recent
 +1 point
+1 point
The JC polyomavirus (JCPyV/JCV) is a member of the Polyomaviridae family and is ubiquitious in the general population, infecting 50–80% of individuals globally. The virus remains latent but can reactivate under conditions of immunosuppression and cause life-threatening disease such as progressive multifocal leukoencephalopathy (PML). PML can be a complication of HIV disease especially in HIV patients who are not receiving anti retroviral therapy. In immunocompetent individuals, PML is rare however, the incidence of PML is increasing due to the widespread use of immune modulating therapies. JCV induced PML is rare in solid organ transplant patients but there is documented cases that correlate with immunosuppression that is required allograft transplantation. Currently there no curative therapies for PML with high mortality after diagnosis.
The JC polyomavirus (JCPyV/JCV) is a member of the Polyomaviridae family, genus Orthopolyomavirus, which also includes the BK polyomavirus (BKPyV/BKV)[1][2][3]. Both viruses were isolated from patients with the initial JC and BK polyomaviruses, respectively[1][2]. JCV is thought to be transmitted by inhylation and infects 50–80% of the global population[4][5], with 10–30% of the world population shedding JCV in their urine[6]. JCV produces an asymptomatic, persistent infection primarily in the renourinary tract, reactivating only when the immune system is compromised. The reactivation of latent JCV in the brain of immune-compromised individuals may result in a lytic infection of oligodendrocytes, glial cells, and astrocytes[7][8][9][10][11]. The demyelination of oligodendrocytes induced by JCV lytic replication may lead to progressive multifocal leukoencephalopathy (PML). PML is a frequently fatal demyelinating infectious disease of the brain caused by neuropathogenic prototypes of JCV and is accompanied by progressive neurological deficits and death[11][12][13][14][15]. The spectrum of neurological presentations includes ataxia, hemiparesis, hemianopia, cognitive impairment, limb ataxia, gait disorder, and aphasia. The commonly involved areas include the subcortical white matter, periventricular areas, and cerebellar peduncles. PML is rare, with an incidence of <0.3/100,000 persons a year among the general population[16]. However, the incidence of PML HIV/AIDS patients without antiretroviral therapy is 2.4 per 1000 persons a year[17]. PML occurs in 3–5% of HIV-infected individuals with an uncontrolled disease, but HIV therapy has been shown to improve clinical outcomes[6][10][11][18]. This is a significantly higher incidence when compared to patients with other means of immunomodulation therapy. The clinical presentations of PML disease include motor weakness, speech abnormalities, cognitive deficits, headache, visual fields deficits, ataxia, aphasia, cranial nerve deficits, and seizures[8][10][19].
Currently, no specific treatment exists for JCV-induced PML. This represents an urgent unmet medical need for efficacious therapeutics for JCV-induced PML in immunocompromised patients. In addition, no suitable animal model for PML exists due to the failure of JCV to productively replicate in nonhuman hosts cells[20][21]. Here, we examine the role of iatrogenic immunosuppression on JCV reaction and the neuropathology that may result post-reactivation.
JCV, along with BKV, identified in 1971, were the first two polyomaviruses associated with human, but not oncogenic, disease[2][3]. To date, 13 different polyomaviruses exist that may infect humans, and this number likely will rise due to novel methodologies used in virus discovery[22]. Five out of the 13 human polyomaviruses have been associated with clinical disease, including JCV, BKV, Trichodysplasia spinulosa polyomavirus, human polyomavirus 7, and Merkel cell polyomavirus[22]. JCV is a small nonenveloped, double-stranded, circular DNA virus with a five-kilobase genome associated with cellular histones, forming a viral mini-chromosome that replicates in the nucleus of permissive cells. Erickson et al. showed that the JCV recruits and modulates the host DNA damage response to replicate its genome[23]. JCV encodes six proteins, the large T and small t antigens, which have oncogenic properties, three capsid proteins (VP1–3), and the agnoprotein[24]. Currently, it is unknown whether the transmissible form of the virus is archetypal (found in the kidney, urine, and sewage, such as the CY strain of JCV); prototypical (found in the brain and is associated with PML, such as the Mad-1 strain of JCV); or both. These two types of JCV viruses differ only within their noncoding regulatory region (NCRR). The prototypical JCV-PML types isolated from the cerebrospinal fluid (CSF) and brain tissues from PML patients have rearrangements, including duplications, tandem repeats, and insertions and deletions in the NCRR region. Evidence of JCV persistence has been found in renal tissue, bone marrow, and brain tissue. This state of persistence appears as episomal circular DNA that is nonreplicating, with no viral gene transcription. Evidence of genomic DNA in normal brain tissue has been observed[25]. The asymptomatic shedding of JCV occurs in the adult human population at a level of 50,000 copies/mL of urine. It has been demonstrated by Coleman et al. that pregnant women in their second and third trimesters may shed JCV asymptomatically at a rate of 3.2%[26]. The seropositivity rates for JCV are found in the range from 39% to 81% and to steadily rise as an individual reaches adulthood, having more sustained viral titers when compared to BKV[5][27]. Primary infections are thought to occur in childhood via the upper respiratory tract through inhalation and, also, via the gastrointestinal route due to contaminated food or water (Figure 1). These findings are supported by the presence of JCV in the tonsils and intestinal cells, respectively[28][29][30]. In addition, JCV is known to infect B cells in the peripheral circulation, adding to the complexity of viral transmission (Figure 1). JCV infection is endemic and ubiquitous, and transmission occurs in childhood by the feco-oral, urino-oral, and respiratory routes. The virus will remain latent in the bone marrow, kidneys, tonsils, lymphoid tissues, and/or the brain throughout the life of an infected individual (Figure 1). Over time, if an individual becomes immunocompromised, JCV may reactivate from latency and cause a life-threatening clinical disease, most notably in the form of PML (Figure 1).
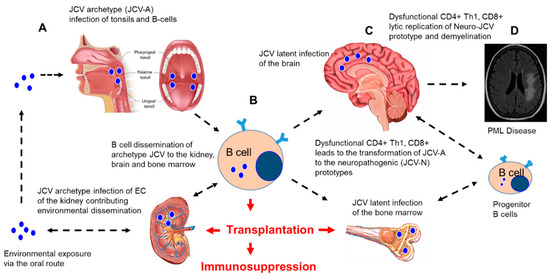
Figure 1. JC polyomavirus (JCV) infection and dissemination from environmental exposure to a latent infection and implications for immunosuppression after transplantation. A hypothetical model of JCV infection, latency, and reactivation in the brain after iatrogenic immunosuppression in transplant patients. (A) Environmental exposure to JCV occurs via the oral route with the initial infection and replication of the non-neuropathogenic archetype (JCV-A) in the tonsils and B cells. (B) JCV-A infection of B cells leads to the dissemination of JCV-A to the kidney, bone marrow, and brain, where latent infections are established. (C) Immunosuppression for a solid organ or stem cell transplantation results in the suppression of immune surveillance, resulting in the impairment of CD4+, Th1, and CD8+ responses and the infection of B cell progenitors that may lead to transformations (mutations in the noncoding regulatory region (NCRR) and VP-1 capsid protein) of the JCV-A prototype virus to the neuropathogenic prototype virus (JCV-N) that induces lytic replication in the oligodendrocytes and demyelination. (D) Uncontrolled multifocal lytic replication of JCV-N in the brain, followed by progressive demyelination, resulting in progressive multifocal leukoencephalopathy (PML), as diagnosed by brain imaging. Figure 1A was modified from a figure provided by Teach-Me-Anatomy. Figure 1C was modified from an image developed by Tim Taylor from Inner Body Research, Brain: Anatomy Mastery Course. Figure 1D was obtained and modified from Infectious Disease Advisor: JC polyomaviruses: Progressive Multifocal Leukoencephalopathy (Clinical Condition). The bone marrow image was obtained and modified from Health Visions, bloodless bone marrow transplants Narayana Health City. We acknowledge the National Kidney foundation for the image of the human kidney (unpublished data) EC: endothelial cells.
JCV is the only human neurotropic polyomavirus that reactivates in the brain under conditions of immunosuppression to cause a severe life-threatening clinical disease. JCV latency is defined as a subclinical, chronic, persistent infection, where JCV DNA, not proteins, may be detected at the site of latent reservoirs. This occurs after immune control of the viremic phase. Polyomavirus-associated nephropathy typically occurs due to BKV, but on occasion, it occurs due to the reactivation of JCV, which occurs almost exclusively in immunosuppressed individuals. JCV rarely causes kidney disease, whereas BKV is a known cause of viral nephropathy[31]. There is a higher incidence of BKV reactivation than JCV reactivation in renal transplant patients[32]. BKV reactivation, as demonstrated clinically by active viruria, which occurs in 23–57% of renal allograft recipients, and BKV-associated nephropathy in as many as 8% of renal allograft recipients. In a study by Lopez et al., they estimated the incidence of JCV viruria to be around 25%[33]. Wiegley et al. observed that, unlike BKV infection, which occurs early post-transplantation, JCV nephropathy occurs at both early and late times after renal transplantation, and that differential diagnosis for BKV may be problematic[34].
JCV often establishes latency in the kidneys, where it displays a stable archetypal noncoding control region (NCCR) in the noncoding region of its genome (Figure 1). The rearrangement of gene sequences in the NCCR of JCV DNA is essential for the reactivation of these latent forms that are associated with PML[35]. The primary sites of viral latency after transmission include the kidney, bone marrow, and B lymphocytes (Figure 1)[36][37][38]. The virus enters the brain via circulation by penetration of the blood–brain barrier (BBB) to establish a nonproductive/low level of infection in the glial cells[39]. No clear understanding of JCV reactivation has been established; however, it has been suggested that a high incidence of infection and the rare incidence of PML imply that many host cell factors are involved in suppressing JCV reactivation[40] (Figure 1). The immune system plays a key role in controlling JCV reactivation from latency, leading to active viral replication and the development of PML (Figure 1). It has been proposed that transcription factor-binding sites located in the NCRR induce cytokine signaling transduction pathways in the binding factors, such as NF-kB, AP-1, Egr-1, and C/EBPβ, which serve to jumpstart viral transcription and DNA replication in response to extracellular cytokines, with primary lesions developing at local sites in the brain parenchyma[41][42][43][44][45]. Further progression of the lesions occurs in the absence of CNS immunosurveillance during immunosuppression. In HIV-infected patients who develop PML, it is proposed that the HIV Tat protein has a role due to its ability to enhance JCV transcription and viral replication [46][47]. B cells are implicated in harboring and trafficking latent prototype PML-JCV in the CNS[48]. The exact viral and host cell factors that contribute to JCV reactivation are currently unknown; however, the outcome appears to depend on the nature of the immunosuppression and patient-specific factors that predispose individuals to JCV reactivation in the CNS and the development of PML disease.





