1000/1000
Hot
Most Recent
 +1 point
+1 point
In the 21st century, enteric fever is still causing a significant number of mortalities, especially in high-risk regions of the world. Genetic studies involving the genome and transcriptome have revealed a broad set of candidate genetic polymorphisms associated with susceptibility to and the severity of enteric fever. This entry attempted to explain and discuss the past and the most recent findings on human genetic variants affecting the progression of Salmonella typhoidal species infection, particularly toll-like receptor (TLR) 4, TLR5, interleukin (IL-) 4, natural resistance-associated macrophage protein 1 (NRAMP1), VAC14, PARK2/PACRG, cystic fibrosis transmembrane conductance regulator (CFTR), major-histocompatibility-complex (MHC) class II and class III. These polymorphisms on disease susceptibility or progression in patients could be related to multiple mechanisms in eliminating both intracellular and extracellular Salmonella typhoidal species.
Enteric fever is caused by the Gram-negative bacillus, Salmonella typhoidal species namely S. enterica serotype Typhi which is responsible for typhoid fever and S. enterica serotype Paratyphi which results in paratyphoid fever, and is considered as a major worldwide health problem [1][2][3]. The disease is characterised by prolonged fever, generalised fatigue, headache and anorexia. In general, the outbreak of enteric fever mainly results from faecal–oral transmission through the ingestion of food and water contaminated with human excreta [4][5]. Hence, the risk factors associated with enteric fever could be related to poverty and social inequality, e.g., through lack of sanitation and hygiene, human demographics and behaviour, as reported elsewhere [6][7].
The occurrence of typhoid fever extends throughout the globe. It is prevalent primarily in developing countries with poor sanitary conditions. Typhoid fever is native to Africa, the Caribbean, Asia, Oceania and Latin America. However, most of the cases originate from Laos, Vietnam, China, Pakistan, Bangladesh, Nepal, India and Indonesia [8]. Within these nations, typhoid fever is mostly reported in underdeveloped regions. According to the model-based estimation carried out by Antillón et al., in 2015, approximately 17.8 million of typhoid fever cases per year are estimated in people who are living in low and middle-income countries [6].
In addition, the ‘global burden of disease 2016′ project shows that there is a slight decrease in the global age-standardised typhoid fever death rate, which dropped by 21.1% from 2.155 deaths per 100,000 in 2006 to 1.7 deaths per 100,000 in 2016. Furthermore, paratyphoid fever deaths decreased by 6.6% from 137.3 thousand deaths to 128.2 thousand deaths in 2016 [9]. Although enteric fever cases declined in a number of countries in recent years, there were still high incidences of typhoid fever in some regions in Africa, as estimated by the adjusted incidence rate, which indicated that children in the age range of 2–14 years have the greatest typhoid fever burden [10]. Importantly, since the year 2000, its increasing resistance to ciprofloxacin treatment has also attracted considerable attention.
Typhoid vaccines are developed to protect individuals from enteric fever. One of them is an injectable polysaccharide (PS) vaccine (also known as ViCPS vaccine), an inactivated subunit vaccine composed of long chains of sugar molecules that make up the surface capsule of S. Typhi. Another vaccine is an edible vaccine which was also developed from live attenuated mutant strains of S. Typhi Ty21a. However, not all vaccine recipients receive adequate protection and benefits from the immunisation against this intracellular Salmonella spp. It is believed that the genetic variation of the recipients plays a major role in modulating the antibody response to typhoid vaccine [11]. They found that alteration of genes involved in PS recognition, signalling ligands and receptors are the main genetic polymorphisms associated with typhoid vaccination outcomes. In addition, genetic variation also influences the individual’s susceptibility to enteric fever. For example, PARK2/PACRG polymorphism is found to be among the candidate gene variants that are greatly associated with enteric fever [12]. In this review, we summarise the most recent and some past findings on human genetic variants affecting biological functions in influencing the outcome of Salmonella typhoidal species’ infection. Table 1 lists the influence of human genetic factors in the development of enteric fever and the role of the genes are illustrated in Figure 1.
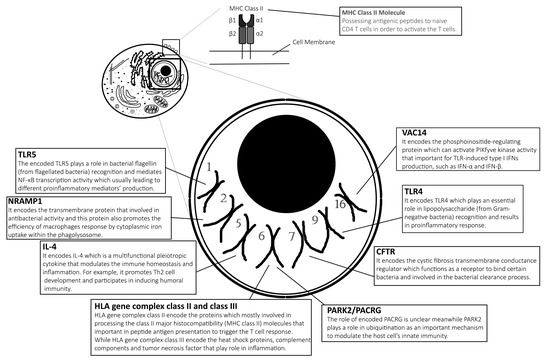
Figure 1. Genes responsible for diverse biological functions. Alteration of the specific genes might affect the susceptibility or severity to Salmonella typhoidal species infection as described and discussed in the content of this review. Notes: This illustration of the eukaryotic cell does not reflect its actual features, while it is just purely for illustration and only seven pairs of chromosomes are represented in this diagram.
Table 1. The influence of gene variants on enteric fever.
| Gene a | Location of Gene | Influence of Variants | Reference |
|---|---|---|---|
| Gene Variants that Affect the Enteric Fever Outcomes | |||
| TLR4 | Chromosome 9 | Missense mutation in TLR4 (threonine → isoleucine substitution at position 399 of the amino acid sequence) or (aspartate → glycine substitution at position 299 of the amino acid sequence) is evidenced to be associated with an increasing risk for Salmonella infection and severity of enteric fever. | [13][14] |
| IL-4 | Chromosome 5 | Variable number of tandem repeat polymorphisms of 3R2R at IL-4 could be a genetic predisposition factor for S. Typhi or S. Paratyphi infection. | [15] |
| VAC14 | Chromosome 16 | The polymorphism rs8060947 in VAC14 gene renders it susceptible to infection. | [16] |
| PARK2/PACRG | Chromosome 6 | Mutation in PARK2 results in a single-nucleotide polymorphism of PARK2_e01(−2599) which shows the weak association and susceptibility to typhoid fever and paratyphoid fever. | [12] |
| CFTR | Chromosome 7 | Polymorphic dinucleotide repeats in the intron or exon of the CFTR gene and also in the single nucleotide variant, whereas, polymorphisms poly-T at CFTR gene are found to be associated with protection against enteric fever. | [17][18] |
| HLA Gene Complex Class II and Class III | Chromosome 6 | HLA-DRB1*04:05 and TNF*1 (−308) allele is associated with resistance to enteric fever whereas HLA-DRB1*0301/6/8 and HLA-DQB1*0201-3 allele are associated with susceptibility to enteric fever. TNFA*2 (−308) is associated with the outcome of Salmonella typhoidal species infection. |
[19][20][21] |
| Gene Variants that Do Not Affect the Enteric Fever Outcomes | |||
| TLR5 | Chromosome 1 | TLR5 variants do not have a significant effect on the susceptibility or severity of enteric fever. | [22][23] |
| NRAMP1 | Chromosome 2 | NRAMP1 polymorphisms are not associated with acquiring enteric fever. | [24] |
a The studies were conducted at the endemic regions of the countries/continent (such as Malaysia, Vietnam, Africa, India, Indonesia and Netherlands) for each polymorphism.
It is well known that Toll-like receptors (TLRs) are used by many innate immune cells to recognise the microbes upon binding to its pathogen-associated molecular patterns (PAMPs), such as components of microbial membranes, cell walls, proteins and oligonucleotides. This interaction complex may lead to the activation of innate immune cells in promoting the production of pro-inflammatory mediators and inducing an appropriate expression of receptor molecules, such as costimulatory molecules, cytokine or chemokine receptors and integrin molecules. Altogether, the activation of TLRs in innate immunity is also one of the crucial steps to initiate the subsequent adaptive immune response which has been elaborately reviewed by others [25][26][27][28][29][30][31]. In short, the activated innate immune cells, particularly dendritic cells (DCs) secrete an array of cytokines which include the interleukin-6 (IL-6), that allows effector T cells to subdue the suppressive effect of regulatory T cells. Collectively, these mechanisms mediate the activation of T helper (TH)-cell immunity and induces the maturation of B-cells which are responsible for the clearance of bacteria [32][33][34][35]. In the case of TLR4 activation, upon lipopolysaccharide (LPS) binding, it bestows the early mounting of nonspecific immune response to infections. For S. Typhi, the TLR4 not only recognises the LPS molecules, but also the porin expressed on the outer membranes of the bacterial cell wall. Cervantes-Barragán et al. found that S. Typhi OmpC and OmpF porins were important to provide signalling via TLR4, to activate the innate immune cells and B cells in enhancing the S. Typhi porins-specific antibodies production [36]. The more detailed description about the immune response after the infection by S. Typhi or other species of Salmonella may be referred through reviews elsewhere [35][37][38][39].
Thus, it is not surprising that polymorphisms in TLR4 might cause predisposition to susceptible infection by S. Typhi. Several other studies have reported that mutation in TLR4 gene or absence of this gene increases the susceptibility of S. Thyphimurium in mice, which mimics human typhoid infection [13][31][40][41][42][43][44][45][46]. Poltorak et al. and Hue et al. identified that the TLR4 gene mutation causes defective TLR4 signalling [29][44], which might result in a lack of inflammatory cytokine production and decrease in the shaping of an adaptive immune response [47]. TLR4 Asp299Gly is a missense mutation that causes the substitution of the aspartic acid residue with glycine at amino acid 299 in the fourth exon of TLR4, whereas TLR4 Thr399Ile is a mutation that occurs by the substitution of a non-conserved threonine residue with isoleucine at amino acid 399 [30][31][48]. Polymorphisms at these amino acids alter the PAMPs-binding site of the extracellular domain of the TLR4 receptor and enhance the susceptibility to enteric fever [13][30][31]. Arbour et al. found that the individual with both TLR4 Asp299Gly and TLR4 Thr399Ile polymorphisms would be hyporesponsive when challenged with LPS and was less efficient in eliciting distinct innate immune responses [31]. The polymorphic variant of TLR4 also results in increased susceptibility to other microbial infections, such as Toxoplasma gondii, Cytomegalovirus, Neisseria meningitides, Mycobacterium leprae and other intracellular pathogens [14][49][50]. However, further discussion about this TLR4 polymorphism association with non-Salmonella infectious diseases is not within the scope of this review. We suspect that the polymorphisms in TLR4 might result in intracellular killing mechanisms to be affected. To the extent of our knowledge, intracellular TLR4 signalling is able to increase the release of nitric oxide within the phagocytes [45][51], and hence mutant TLR4 alleles may result as ineffective in bactericidal activity on intracellular S. Typhi by nitric oxide.
In a study performed on a Malay population in Malaysia, the significantly higher frequency of the mutant G (299 Gly) allele and mutant allele T (399 Ile) in susceptible individuals, as compared to that in the healthy controls, showed an association between these mutant alleles with typhoid susceptibility. The genotype frequency of TLR4 Thr399Ile heterozygous variant was approximately two-fold higher than in healthy controls. On the other hand, the mutant G allele at the Asp299 locus was approximately three times higher in the typhoid susceptible group than in the control subjects [13]. Bhuvanendran et al., reported that the prevalence of TLR4 Asp299Gly and Thr399Ile polymorphisms was 8.9% and 7.2%, respectively, in the typhoid susceptible population, which compares to 1.8% and 3.6% in the corresponding normal population. The co-segregation of these alleles was observed in 2% of normal controls and 3.6% in typhoid susceptible individuals. However, another study demonstrated that monocytes from Asp299Gly heterozygotes did not influence the ability of monocytes in LPS recognition, where the activation of TLR4 downstream signalling still occurs [52]. This finding is in contrast with the data obtained from different research groups as discussed above. For example, Arbour and colleagues reported that airway epithelial cells obtained from individuals heterozygous for the TLR4 mutations did not respond to LPS stimulation [31]. This highlights the complexity of the TLR4 system in different cells and tissues. The detailed mechanisms and evidence on different immunological aspects of the TLR4 system at distinct cell types are poorly understood. Furthermore, differences in immune responses have not been evaluated with other TLR4 ligands, such as purified porin.
Other than in a Malay population, there were 13 TLR4 polymorphisms reported in a study done among Caucasian and Dutch populations [53][54]. TLR4 Asp299Gly mutations are common variants with a frequency of >10% in the Caucasian population and as high as 21.5% in Ghanaian Africans [55][56]. TLR4 Asp299Gly and TLR4 Thr399Ile polymorphisms commonly co-segregate as well as inherit together in European whites but not in African population [57][58][59]. The lack of co-segregation for Asp299Gly and Thr399Ile alleles in the African population could be one of the reasons for the higher incidence and mortality rates of enteric fever. This is because a study had indicated that the presence of the TLR4 Asp299Gly allele alone is associated with an increased severity of Gram-negative bacterial infection [46].
Despite the common functional TLR4 mutants (Asp299Gly and Thr399Ile) found in Malaysia, Europe and Africa, it came as a complete surprise that both these alleles were reported to be absent or present at a low frequency in the Vietnamese population, especially TLR4 Asp299Gly [31][44]. In contrast, DNA sequencing studies performed on a Vietnamese population observed that the exonic polymorphisms actually related to the mutations in the N-terminal leucine-rich repeat (LRR) region within the extracellular domain of TLR4, when compared to reference sequences (NCBI accession number AF 177765) [44]. These mutations in the LRR region, particularly TLR4 Ser73Arg, showed significant association with enteric fever because it could disturb the phosphorylation of TLR4, thus altering the downstream signalling of an inflammatory mediator activation. However, the question of whether the TLR4 Ser73Arg could be the main SPN effect on typhoid susceptibility if this study repeated in large sample size is still not known yet.
Collectively, the data obtained from molecular genetic studies indicated that the presence of rare missense mutations in the TLR4 gene, particularly those that influence the extracellular domain of TLR4, may affect the immune response to enteric fever; and the polymorphism may also be different according to the varying ethnic backgrounds. However, it is currently unknown whether there is any influence on the new functional TLR4 mutants on the host response to S. Typhi infection. To date, there have not been many studies on TLR4 polymorphisms in a larger geographic range for the S. Typhi infected patients that would give rise to clinical diversity. Other than TLR4, TLR2 polymorphism and its relationship with typhoid fever have also been studied. Only one such study led by Sivaji’s team at Tamil Nadu, India, in 2016, showed that the TLR2 polymorphism induced susceptibility to typhoid infection since it was found in 10% (2/20) of typhoid patients. This TLR2 acts as a receptor that recognises LPS and may also recognise lipoproteins in order to trigger a strong immune response.
TLR5 is believed to have the ability to recognise the flagellin of S. Typhi and thereby stimulate an immune response similarly to in TLR4 activation, which had been evidenced via animal studies [26][33][60][61]. The availability of functional TLR5 is needed to play an important role against flagellated bacterial infection [62][63]. For example, various polymorphisms in the TLR5 gene by introducing a premature stop codon (TLR5392STOP) within the PAMPs binding domain are correlated to different degrees of interleukin 6 (IL-6) production, which may affect innate and adaptive immune responses [64][65]. It has been reported that single nucleotide polymorphisms (SNPs) in TLR5392STOP are associated with Legionnaires’ disease caused by Legionella pneumophila [64]. However, this would not be the same story for enteric fever. In recent years, experiments have been carried out to investigate the relationship between the host susceptibility to enteric fever and the loss of TLR5 or its polymorphism TLR5 on chromosome 1. Surprisingly, most of their findings showed discrepancies from the study focused on TLR4 polymorphisms as discussed above. They showed that the variation in the TLR5 gene did not affect individual susceptibility to typhoid fever [22][66]. For instance, Senthilkumar et al. reported that the TLR5392STOP polymorphism did not show any association with clinical manifestations in Indian patients with typhoid fever and asymptomatic typhoid carriers. Before that, Dunstan et al. had already analysed the stop codon polymorphism of TLR5 in patients with enteric fever compared to healthy Vietnamese individuals (565 patients; 281 controls) for the samples collected between 1995 and 2002 [22][23][66]. Similarly, this study also did not find any significant difference between both studied groups. Therefore, it could be postulated that a mutation at the PAMPs domain binding site of TLR5 (individuals with the TLR5 stop codon) might have no correlation with susceptibility to enteric fever progression.
Natural resistance-associated macrophage protein 1 (NRAMP1) is a transmembrane protein found in the endosomes and lysosomes of monocytes and macrophages, which are encoded by the NRAMP1 gene (also called SLC11A1). This plays an important role in exhibiting its antimicrobial activity and is also involved in iron homeostasis for macrophages to enable macrophage functions properly [67]. Variations in NRAMP1 gene at chromosome 2 is therefore expected to cause a broad range of susceptibility to infection [1][68][69]. Interestingly, a recent study by Cunrath et al. unravelled that NRAMP1 could decrease the growth of nontyphoidal Salmonella by limiting the magnesium availability for Salmonella as shown in their in vivo assay [70]. Nevertheless, polymorphisms within the NRAMP1 gene are not expressed in correlation with typhoid fever. According to Dunstan et al., there were no allelic associations that were identified among the NRAMP1 alleles and typhoid fever susceptibility [24]. Therefore, none of the homozygotes and heterozygotes of the NRAMP1 variants are at increased risk of typhoid fever. However, some studies showed that NRAMP1 polymorphisms may be associated with infectious diseases such as tuberculosis (TB), leprosy, rheumatoid arthritis and Crohn’s disease [68][69][71][72]. According to Medapati et al., 3′-UTR, INT4, D543N and 5′-(GT)n polymorphisms of NRAMP1 were significantly associated with TB among west Africans [73]. Another study conducted by Brochado et al. reported the 274C/T polymorphism in exon 3 and the 469 + 14G/C polymorphism in intron 4 of the NRAMP1 gene which was found to be associated with susceptibility to leprosy [74].
VAC14 is a phosphoinositide-regulating protein which activates PIKfyve kinase activity by forming a regulatory complex with PIKfyve and the gene that encodes VAC14 is located on chromosome 16 [75][76]. PIKfyve, a class III lipid kinase, is crucial in TLR-induced type I IFN production. Although the polymorphisms of TLR5 and NRAMP1 do not show impact on enteric fever susceptibility, S. Typhi clearance is currently reported to be influenced by VAC14 variations [16]. One of the more serious SNPs of the VAC14 gene is an allele of rs8060947, which is strongly associated with susceptibility to S. Typhi invasion and decreased VAC14 expression in Vietnamese individuals [16]. There is another study which demonstrated that the variation in VAC14 for Kenyan children could also be readily associated with the increased risk of bloodstream infection from Escherichia coli, Acinetobacter spp., nontyphoidal Salmonella as well as Gram-positive Streptococcus pneumoniae [77][78]. A genome-wide association study (GWAS) complemented with high-throughput human in vitro susceptibility testing (Hi-HOST) showed the mutations in the VAC14 gene and it has been linked to an increase in plasma membrane cholesterol which in turn facilitates the Salmonella Typhi docked firmly to the host cell [16]. There is plenty of literature showing that plasma membrane cholesterol content is an essential factor for E. coli, S. pneumoniae and toxins of Acinetobacter species to enter the host cells [79][80][81]. Therefore, the enhanced expression of VAC14 could be responsible for its protection against S. Typhi and suggests that rs8060947 could be a biomarker for enteric fever disease risk prediction.
As mentioned above, VAC14 is also one of the essential protein subunits for PIKfyve in signalling the type I IFNs production. Therefore, the impact of VAC14 allele of the variant rs8060947 might interrupt the cells in producing type I IFNs which can either be beneficial or otherwise to the host defence system. However, polymorphisms of VAC14 in relationship with type I IFNs production is yet to be investigated for the S. Typhi infection models [82]. In general, type I IFNs comprise IFN-α and IFN-β, in which are both important in antiviral response; but with abnormal upregulation, may lead to the progression of systemic lupus erythematosus (SLE). Until recently, IFN-β has been reported as an important cytokine that stimulates the prevention of the dissemination of S. Typhimurium and enhances Salmonella clearance after the host has been treated with polyinosine–polycytidylic (poly I:C) [83]. A notable feature of the IFN-β is that it also acts as an inducer by enhancing the IFN-γ production from CD4+ cells, especially the TH1 cells [38].
It is well known that IFN-γ plays a critical role in innate immunity as well as adaptive immunity [84][85]. Previously, IFN-γ was detected in high titer in patients with S. Typhi bacteremia as reported by Sheikh et al. [86]. Of note, Bhuiyan and colleagues also demonstrated the increased IFN-γ response after stimulation with different typhoid antigens and this was consistent with the findings of Sheikh et al. [38]. Other than that, the increased IFN-γ response was observed in a subject from Nepal who was infected with S. Typhi and S. Paratyphi A, while there was a report which showed that a child with a complete loss of IFN- γR1 was killed by S. Typhi infection [87][88]. The role of IFN-γ has been evaluated by Nairz, who showed that IFN-γ could reduce the iron uptake of macrophage through transferrin receptor [89]. Hence, in other words, it increased the efflux of iron; and at the same time, strengthened the ability of macrophages to kill intracellular pathogens including Salmonella [90]. The bacteria cannot grow well in the absence of iron. The IFN-γ also induces the expression of other proteins (such as NRAMP1 and lipocalin 2) that are involved in the reduction in cytoplasmatic iron or other divalent cations in the infected host cells [91][92]. Other roles of IFN-γ such as phagolysosomal maturation, oxidative and nitrosative burst, autophagy and decreasing the levels of tryptophan to reduce the growth of intracellular Salmonella can be observed in the literature [51][82][93][94][95][96].
Variable number of tandem repeats (VTNRs), also known as single-copy mini-satellites, consists of consecutive occurrences of nucleotide sequences at a particular locus [97]. VNTRs sequences play a major role in regulating the transcription, translation and function of proteins when located within the coding area of certain genes. This, in turn, could lead to severe forms in different diseases [97]. The mode of how VTNRs induce changes in IL-4 expression, which is located on chromosome 5, will be described in this section. Generally, VNTR polymorphisms in IL-4 intron 3 often occur in four different ways; allele with two repeats (2R), three repeats (3R), four repeats (4R) and rarely with one repeat (1R) [15]. Therefore, these mutations seem to be associated with susceptibility to and/or progression towards enteric fever upon S. Typhi or S. Paratyphi infections [98]. In fact, there are not many studies on the association of VNTR IL-4 polymorphisms with other infectious diseases. We only acknowledge that some VNTR IL-4 polymorphisms could affect the host susceptibility to brucellosis which was caused by intracellular Brucella melitensis [99]. As far as we know, any VNTR polymorphism at IL-4 intron 3 is also associated with other immunological diseases, such as rheumatoid arthritis, immune thrombocytopenic purpura, systemic lupus erythematosus, multiple sclerosis and knee osteoarthritis [98]. This is because IL-4 is one of the major cytokines responsible for both innate and adaptive immunity which influences various cell types. It has been demonstrated that the polymorphism of 3R2R in IL-4 most likely results in the reduction in protection against S. Typhi and S. Paratyphi, conferring a higher risk in the development of enteric fever [15]. In contrast, the polymorphism of 3R3R, 3R1R, and 2R2R does not contribute to any susceptibility towards enteric fever [15]. It was suggested that the IL-4 VNTRs polymorphism might affect the transcriptional activity, including the higher expression of IL-4, leading to a shift in the Th1/Th2 balance toward Th2 [15][100]. It is well known that the protective immune response against S. Typhi is the maturation of Th1 cells, apart from the B cells [38]. Immunisation studies with iron-regulated outer-membrane proteins of S. Typhi have also shown that the protection against Salmonella infection was initiated by the Th1 cells [101]. This is because the Th1 cells may drive macrophages to M1 polarisation [102][103]. It is well known that the activated M1 macrophages play a main role in killing the intracellular pathogen [104]. Through this mechanism, individuals with VNTR polymorphism in IL-4, especially, the 3R2R genotype, are more susceptible to S. Typhi or S. Paratyphi infection.
PARK2/PACRG genes are located on chromosome 6. The PARK2 gene is responsible for encoding ‘parkin’, which plays a major role in ubiquitination [105]. The specific function of PACRG gene is still unclear. Polyubiquitination by parkin for the targeted protein is commonly needed to initiate the proteasome-mediated protein degradation pathway [106]. Both ubiquitination and degradation might play important roles in many biological functions, and the failure of these processes could ultimately lead to many human diseases which are not limited to only non-infectious diseases. Studies have found an association between this protein degradation pathway and the ability of Salmonella spp. to invade the host cells [107][108]. For example, upon contact with the host cells, Rho GTPases Cdc42 and Rac1 in the host cell are activated by Salmonella effectors encoded by sopE and sptP during the entry process. In general, SopE resulted in extensive membrane ruffling and the actin cytoskeletal network reorganisation of the host cell which is important for bacterial uptake into host cell, while SptP restores the host cell architecture. The SopE can be rapidly degraded while SptP is slowly degraded within the host cell via an ubiquitin/proteasome-mediated degradation pathway. Therefore, any mutation in PARK2 that alters its ubiquitination ability will undoubtedly affect the half-life of SopE. Consequently, this inhibits cellular recovery after bacterial infection, and results in persistent membrane ruffling which allows the S. Typhi or S. Paratyphi to grow continually inside the host cells as it easily enters the cells [109][110].
As aforementioned, parkin encoded by PARK2 was observed to play a role in ubiquitination, while most importantly, this mechanism also modulates the host cell’s innate immunity. Ubiquitination is an essential part in innate immunity by signalling the cells for the proper secretion of various signalling mediators [111]. Up to now, it is believed that parkin might act as a mediator for the production of two key pro-inflammatory cytokines: IL-6 and monocyte chemoattractant protein 1 (MCP-1). In this, IL-6 contributes to systemic inflammatory response or differentiate the activated B cells, and MCP-1 attracts monocytes into the infected area. This has been proven by the downregulation of the PARK2 gene expression in Schwann cells, monocyte-derived macrophages and THP-1 macrophages [112]. Moreover, it has also been shown that polyubiquitination contributes to autophagic pathway by coating the intracellular pathogen with ubiquitin which enables it to be readily targeted for autophagy. Details regarding the autophagy in the host cell’s innate immunity can be reviewed in other literature [113][114][115]. Indeed, a few in vitro and in vivo studies have been performed to elucidate the functional mechanism of the PARK2 gene in autophagic immunity against M. tuberculosis, S. Typhimurium and Streptococcus pyogenes [116][117][118]. The expression of PARK2 was observed to recruit more ubiquitin and ubiquitin-binding autophagy adaptors to the bacteria, which resulted in the reduction in bacterial survival, whereby increasing the infected cell survival. Therefore, it is not surprising that there might be an association between PARK2 alleles and innate immune response. Notably, the polymorphisms of PARK2/PACRG genes were found to be associated with susceptibility to infectious diseases including typhoid and paratyphoid fever [12][105][119][120]. Additionally, different variants of PARK2/PACRG genes were also reported in association with susceptibility to infection caused by M. leprae, for example, SNP of rs1333955, rs10806768 and rs9355403.
In the last decade, a case-control study was conducted in the Jatinegara district of Jakarta, Indonesia. This study examined the polymorphisms in PARK2/PACRG genes at the shared 5′ regulatory region of PARK2 and PACRG. This was performed to study both the environmental factors and genetic determinants towards the susceptibility to enteric fever. Four SNPs (PARK2_e01(−2599), PARK2_e01(−697), rs1333955 and rs1040079) were used for this study in evaluating the polymorphic variants of the PARK2/PACRG genes. This was carried out in 116 enteric fever patients, with 337 as fever controls and 332 as community controls. The findings showed genetic variations at this targeted gene region, which might be correlated with the individual susceptibility to enteric fever. By combining the results of Hardy–Weinberg equilibrium of each SNPs and association study, only one SNP (allele T of PARK2_e01(−2599)) among the 4 SNPs analysed was found to be significantly—but weakly—associated with enteric fever (OR: 1·51; CI: 1·02–2·23, p = 0.03). Thus, the mutation in PARK2/PACRG genes, especially the variant PARK2_e01(−2599) has been suggested as one of the risk factors in the development of enteric fever and leading to the prolonged half-life of Salmonella effectors protein [12].
The CFTR gene found on human chromosome 7 encodes the cystic fibrosis transmembrane conductance regulator (up to 1480 amino acids) which functions as a protein channel throughout various types of glandular cells for the secretion of enzymes, sweat, mucus, saliva and tears [121]. Mutations in this gene can result in a lethal genetic disorder known as cystic fibrosis [122][123][124].
Interestingly, CFTR protein also functions as a receptor for type IVB pili of Salmonella Typhi in order to adhere to the intestinal mucosa cells which facilitates the bacterial translocation process into the intestinal submucosa [125]. Afterwards, S. Typhi might induce more intestinal epithelial cells to express its CFTR protein and this could be an essential step for the development of typhoid fever [126]. An in vitro study using isogenic cells has demonstrated that the mutant with phenylalanine deleted at residue 508 (∆508 or F508del mutation) is less susceptible to S. Typhi infection as compared to cells expressing wild-type CFTR [125]. Therefore, F508del mutation is believed to render a selective advantage against S. Typhi or S. Paratyphi infection to the host [17]. Another study using an animal model has demonstrated that the homozygous F508del mice are immune to the S. Typhi translocation into their gastrointestinal submucosa, thereby protected from enteric fever [125]. However, the distribution of F508del mutation in the human populations is uneven throughout the world, and it is uncommon to observe the F508del genotypes in Asian populations. The other form of CFTR mutation could be found to provide a selective advantage against S. Typhi infections.
In years past, CFTR intragenic polymorphic microsatellites analysis indicated that IVS8CA alleles 181 (CA16) and 183 (CA17) on intron 8 of the CFTR gene were significantly associated with resistance to enteric fever and van de Vosse et al. also identified that both IVS8CA genotypes of 181/181 and 181/183 have a protective effect for typhoid fever in a case-control study in Indonesia [17]. In an attempt to identify the contribution of functional mutated CFTR protein to protection against enteric fever, the same research team conducted a study in the Jatinegara district of east Jakarta (Indonesia), by analysing the blood samples positive for enteric fever and blood specimens from healthy control subjects [18]. As expected, the sequence variations in the CFTR gene of the Indonesian population were identified and the distribution of alleles and genotypes of typical polymorphisms were detected between the control subjects and enteric fever patients. However, interestingly, these DNA sequence polymorphisms showed no correlation between the IVS8CA repeat genotypes and susceptibility to enteric fever. In contrast, they revealed that IVS8 TG repeat genotypes are associated with protection against enteric fever, especially individuals who possess one of the CFTR variations or more than one CFTR variations (such as TGn repeat genotype TG11TG12, 2562T > G genotype TG, Q1352H mutation, the alleles of IVS8 TG13 or TG15 and the allele of IVS8 T5) might be more resistant to S. Typhi or S. Paratyphi than other groups in the population [18].
Over the last decade, the major-histocompatibility-complex (MHC) polymorphism has been recognised as a factor determining the various infectious disease susceptibility. MHC molecules are the glycoproteins on the cell surface of eukaryotic cells that play a crucial role in the immune system, autoimmunity and reproductive success [127][128]. The genes that encode this MHC molecule are located within the human leukocyte antigen (HLA) gene complex region either as class I or class II at chromosome 6. Another gene complex that can be found between them is HLA class III which consists of genes encoding tumour necrosis factor (TNF)-α, lymphotoxin (LT)-α and other gene products which do not play the same role as MHC glycoproteins [129]. Variation in HLA regions might result in a different degree of resistance or susceptibility to infectious diseases as discussed elsewhere [130][131][132][133]. In 2001, the polymorphism at HLA class II and class III regions have been extensively investigated to correlate with typhoid fever in the Vietnamese population [19]. According to Dunstan et al., the presences of both TNF*1 (−308) and HLA-DRB1*04 alleles in an individual might result in protection from typhoid fever. After a few years, Dunstan et al., also reconfirmed that variants of the HLA-DRB1 gene are the main key for individual resistance to typhoid fever [20]. The application of imputation-based fine-mapping across the extended HLA region provided a confident datum to show that HLA-DRB1*04:05 allele as a protective factor against typhoid fever with its genotyped single nucleotide polymorphism (SNP), rs7765379. However, the alleles of HLA-DRB1*0301/6/8, HLA-DQB1*0201-3 and TNFA*2 (−308) have been reported to be associated with increased susceptibility to typhoid fever [131][132]. On the other hand, a study had demonstrated that individuals with the allele of HLA-DRB1*12021 were found to be protected from the severe outcomes of enteric fever [134]. The severity of infection was also affected by the mutation at the region of HLA class III, especially the TNFA gene. According to Elahi et al., guanine is substituted by adenosine in the synthesis of TNFA*2 (−308) in TNF-α polymorphism. Thus, the production of TNF-α is stimulated or markedly increased directly due to the presence of TNFA*2 (−308) allele. Consequently, some unwanted immune responses, which include shock, elevated temperature and sleepiness, could be induced by bacterial endotoxin, interleukin-1 and interleukin-6 when the titre of circulating TNF-α is extremely high for the individual with TNFA*2 (−308) polymorphism [135]. Therefore, the TNFA*2 (−308) allele is mostly thought to be associated with the severity of enteric fever instead of susceptibility. Furthermore, in contrast, a study in Vietnam, and a case-control study from Indonesia conducted by Ali and colleagues clearly indicated that there was no association of susceptibility to enteric fever and TNFA polymorphisms, particularly TNFA (−308) [21]. We could speculate that the severity of Salmonella typhoidal species infection might vary significantly between individuals based on TNFA and HLA-DRB1 polymorphisms.





