1000/1000
Hot
Most Recent
 +1 point
+1 point
Suppression of tumorigenicity-2 ST2 has emerged as one of the most promising biomarkers in assessing the evolution and prognosis of patients with HF. The uniqueness of ST2 is determined by its structural particularities. Its transmembrane isoform exerts cardioprotective effects, while the soluble isoform (sST2), which is detectable in serum, is associated with myocardial fibrosis and poor outcome in patients with HF.
One year after its first occurrence, coronavirus disease 2019 (COVID-19) is still dominating not only the public health systems, but also the research spectrum. The current fields of interest are focused on unraveling the immunopathology of COVID-19 caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and assessing the multiple extrapulmonary clinical manifestations related to this new viral infection. Myocardial injury is probably the most severe non-respiratory associated condition in COVID-19 patients and can be elicited by several mechanisms. Recent studies emphasized the presence of a common neuroendocrine pathway, for both pulmonary and myocardial injury, via the angiotensin converting enzyme 2 ACE2 receptors, which are highly expressed in heart and lungs[1].
The internalization of the virus, through binding the ACE2 receptor, can directly cause an initial myocardial injury, as it was previously demonstrated in other severe acute respiratory syndrome SARS viral outbreaks[2]. More recently, the SARS-CoV-2 genome was confirmed by PCR in the cardiac tissue from a recent autopsy study in patients deceased due to COVID19, but without previous clinical signs of fulminant myocarditis. Interestingly, the virus was mostly cantoned in interstitial cells or other inflammatory cells invading the myocardial tissue, and not in the cardiomyocytes themselves[3]. On the other hand, another study highlighted the presence of SARS-CoV-2 preferentially in COVID-19 patients’ cardiomyocytes, both in terms of viral transcript detection, assessment of viral proteins or even virion identification. The same research team also observed the presence of significant injury in infected cardiomyocytes, ranging from intracellular oedema to focal myofibrillar lysis and sarcomere ruptures[4].
Additionally, there is a mutual influence between SARS-CoV-2 and the ACE2 receptor; despite the fact that the virus uses ACE2 to enter the host cell, it also downregulates the expression of this receptor, promoting a cascade of deleterious effects at myocardial level. This is due to ACE2 receptors’ capacity to enhance degradation of angiotensin (Ang) II to Ang 1-7, that presents a cardioprotective capacity via an increased NO release and decreased fibrogenesis[5][6]. ACE2 receptors also limit the negative effects resulting from binding of Ang II to AT1 receptors, which include myocardial hypertrophy and dysfunction, interstitial fibrosis, vasoconstriction, marked proinflammatory status, oxidative stress and hypercoagulability[6][7]. Is noteworthy that positive influence of ACE2 is intimately linked with the upregulation of the renin–angiotensin–aldosterone system (RAAS), as it happens in heart failure (HF), arterial hypertension or atherosclerosis[7]. Basically, the SARS-CoV-2 not only binds the ACE2 receptor in order to enter the cells, but it also downregulates its expression in cardiac muscle, functionally removing it from the external site of the membrane, hence contributing to the poorer outcome of COVID-19 patients suffering from cardiovascular diseases, particularly from HF[8][9].
The pathophysiological chain of events can be summarized as two different molecular pathways, with opposite effects concerning cardiovascular system: the deleterious ACE→Ang II→AT1 receptor pathway that is counter-regulated by the protective ACE2→Ang1–7→Mas receptor pathway[5][6].
ST2 is a subtype of the vaster interleukin 1(IL-1)/ toll-like receptor (TLR) family, also known as interleukin 1 receptor-like 1 (IL1RL-1)[10][11]. Although firstly described in 1989, it was not until the early 2000s that its possible role in cardiovascular system was researched.
The ST2 protein consists of two main isoforms: a transmembrane receptor form (ST2L) and a circulating, soluble receptor form (sST2) that can be detected in serum[11]. The transmembrane isoform ST2L represents the receptor for interleukin-33 (IL-33), which is a functional ligand with extensive expression in different tissues (e.g., epithelial cells, endothelial cells, fibroblasts)[12].
Even if cardiac tissue is considered to be a major expression site, several studies provided evidence that the human myocardium is not the sole source of sST2, increased sST2 mRNA expression also being found in lungs, followed by kidneys, small intestine and brain, with very interesting variations concerning gene transcription between in vivo tissues and in vitro cell cultures[13][14]. Whereas sST2 expression in kidneys was significant in vivo, it was virtually absent in kidney-derived cell cultures. Concerning the specific tissue origin of this molecule, the highest in vitro sST2 mRNA expression was observed in cells derived from lung, with highest sST2 concentration being observed in supernatant containing lung alveolar epithelial cells, lung bronchus epithelial cells, and cardiac myocytes[14].
In order to confirm the intimate cardiopulmonary interconnection regarding sST2 synthesis, Pascual Figal et al. performed an experimental model of HF and observed that sST2 was upregulated in lungs and secreted by type II pneumocytes in response to myocardial strain. Moreover, in patients with cardiogenic pulmonary edema, soluble ST2 is present in high amounts in bronchial aspirates[15].
Specifically, IL-33 is secreted by cardiac fibroblasts in response to myocardial strain or injury[10]. Several experimental studies have proven that the interaction between ST2L and IL-33 promotes cardioprotective effects, by limiting myocardial fibrosis, inhibiting cardiomyocyte hypertrophy, reducing apoptosis and upgrading the overall myocardial function via activation of myeloid differentiation primary response gene 88 (MyD88), interleukin-1 receptor-associated kinase (IRAK), extracellular signal-regulated kinase (ERK) and nuclear factor-κB (NF-κB) signaling pathways[11][16].
Notably, Villacorta et al. observed that cardioprotection occurs exclusively through IL-33 binding to the ST2L, the transmembrane receptor. By contrast, the soluble isoform (sST2) is acting more like a decoy receptor, binding with high avidity to the IL-33 and thus, competing with ST2L and inhibiting the above-mentioned cardioprotective signaling generated by IL-33/ST2L interaction[11]. To make things more challenging, previous studies highlighted that, under biomechanical overload, the ST2 gene is significantly up-regulated in HF; subsequently, cardiac myocytes and fibroblasts steadily release in circulation both ST2L and sST2, whose mutual ligand-IL-33- is also hypersecreted under mechanical strain[11][17]. Additional evidence suggest that knocking-out the ST2 gene or administering high amounts of soluble ST2 to compete for IL-33 will induce a phenotype similar to the one seen in BNP knock-out models, characterized by important myocyte hypertrophy and interstitial cardiac fibrosis[18].
In Table 1 we summarized the data concerning some morphofunctional aspects of ST2 and their impact on cardiac function.
Table 1. ST2: two isoforms with opposite cardiovascular effects.
| Isoform | Site | Ligand | Cardiac Eeffect |
|---|---|---|---|
| ST2L | Cell membrane (expressed in cardiomyocytes, endothelial cells, inflammatory cells) |
IL-33 | Cardioprotection by: -limiting myocardial fibrosis -inhibiting cardiomyocyte hypertrophy -reducing apoptosis |
| sST2 | Bloodstream (soluble isoform, detectable in serum) |
IL-33 | Cardiac dysfunction via inhibition of the above effects |
ST2L—suppression of tumorigenicity-2 ligand; sST2—suppression of tumorigenicity-2 soluble isoform.
Based on multiple previous observations and the assumed role of sST2 in the fibrotic response to myocardial tissue injury, it is important to highlight that the release of sST2 by cardiac fibroblasts and cardiomyocytes is closely related to two profibrotic conditions, very common in HF with reduced ejection fraction (HFrEF): biomechanical strain and elevated Ang II[16][19]. The latter is causing myocardial hypertrophy and fibrosis by enhancing the simultaneous activation of the transforming growth factor β-1 and the connective tissue growth factor via cell signaling, thus leading to an increased synthesis of collagen and other several matrix proteins.
Patients with new-onset dyspnea may represent the starting point for sST2′s diagnosis value assessment in HFrEF. Theoretically, a poor ventricular contractility and/or compliance will consequently increase the myocardial stretch, thus providing a stimulus for both dyspnea and sST2 release. However, connection between an increased serum level of sST2 and systolic dysfunction is marked by controversy in literature data. Evidence supporting this hypothesis is based on the research conducted by Shah et al. who identified a clear association between elevated sST2 and impaired biventricular systolic function (assessed as diminished left ventricle LV ejection fraction and abnormal RV fractional area change)[20]. The same authors also noticed other structural echocardiographic abnormalities commonly found in HFrEF, an increased sST2 concentration being significantly correlated with higher LV end-systolic area and volume. Moreover, Tseng et al. found that patients with HF in NYHA IV functional class present an elevated sST2 compared to those in NYHA II/III, but the serum levels could significantly decrease after the improvement of ejection fraction consecutive to a left ventricular assist device implantation[21].
Although other studies actually indicate that there is no clear association between an elevated sST2 and an impaired systolic function[19][22], they also mentioned the importance of the sST2 in risk stratification of patients with HF, as well as its additive value to NT-proBNP in diagnosing acute HF, both with reduced or preserved ejection fraction.
Data from the Pro-Brain Natriuretic Peptide Investigation of Dyspnea in the Emergency Department (PRIDE) study also highlighted the diagnosis and prognosis value of sST2 in HFrEF, its serum levels being significantly higher in patients with acute HF compared to patients without HF (0.50 vs. 0.15 ng/mL, p < 0.001), and in deceased patients compared to survivors (1.08 vs. 0.18 ng/mL)[23]. Furthermore, the sST2 was also associated with NYHA functional class, LV ejection fraction (LVEF) and BNP/NT-proBNP levels; unlike natriuretic peptides, sST2 was not correlated with age, body mass index, atrial fibrillation or the etiology of HF[11][23]. Additionally, sST2 is presumably a better diagnosis tool in patients with HF and concomitant kidney disease, because the value of ST2 was not affected by renal function, as opposed to NT-proBNP[24].
Data collected from several studies confirm that sST2 is a strong predictor of mortality or HF rehospitalization during follow up, irrespective of other traditional clinical (e.g., NYHA class), biochemical (e.g., natriuretic peptides), or echocardiographic (e.g., ejection fraction) risk markers[11][19][23][25][26]. Another very recent study showed that elevated sST2 levels were superior to NT-proBNP in predicting in-hospital death (95% CI 3.55–16.26, p < 0.001 vs. 95% CI 1.10–3.72, p = 0.037), while another “classic”, widely-used biomarker-hs-cTnI- was not significantly associated with the in-hospital mortality (95% CI 0.28–2.42, p = 0.114)[27]. Under these circumstances, we consider that a multimarker test consisting of natriuretic peptides and sST2 determination will provide incremental diagnostic and prognostic values in patients with HF (Table 2).
| sST2 ↓ | sST2 ↑ | |
|---|---|---|
| NT-proBNP ↓ | 10% | 40% |
| NT-proBNP ↑ | 28% | 56% |
HF with preserved ejection fraction (HFpEF) is characterized by a quasinormal systolic function (LVEF >50%) and concerns specific categories of patients: hypertensive, obese, elderly, women etc. Some authors invoked the presence of a direct relationship between HfpEF and proinflammatory comorbidities, like diabetes, atrial fibrillation or pulmonary hypertension[16].
It is already proven that ST2 is involved in inflammatory and immune processes, with a major role in regulating mast cells and type 2 CD4 T-helper cells, with a consecutive enhanced production of Th2-related cytokines[10]. Other experimental research demonstrated that overexpression of IL-1β, a proinflammatory cytokine, induces ST2 mRNA, especially post-myocardial infarction[17]. The concept of a “bridge” biomarker, suggestive for both increased inflammatory state and HFpEF is furtherly supported by Villacorta et al. who found a correlation between C-reactive protein and sST2 serum level in patients admitted for HF[11].
Regarding clinical status at admission, dyspnea is the most common symptom in patients with HFpEF and represents a marker of congestion, mainly due to increased LV filling pressures, the correlation between elevated sST2 and increased risk of mortality in dyspneic patients being confirmed in several studies[26][28].
Many hypertensive patients meet the criteria for HFpEF, with LV hypertrophy due to poorly controlled long-term hypertension being one of the most common structural findings. Lotierzo et al. reviewed multiple studies and found that increased levels of sST2 are clearly associated with elevated blood pressure, both in patients with or without HFpEF[25].
Diastolic dysfunction and left atrial enlargement represent another significant feature commonly found in HFpEF. Najjar et al. revealed that increased left atrial volumes are correlated with higher serum sST2, a finding inconsistent with previous research that infirmed such an association[19][20]. There is also room for debate concerning LV filling pressures, expressed as E/e’. While some authors claimed that sST2 is significantly higher in patients with E/e’ > 8 [22][29], other studies failed to demonstrate this aspect[19][28].
Another interesting echocardiographic aspect was revealed by Fabiani et al., who observed a LV systolic dysfunction (assessed as global longitudinal strain) in patients with HFpEF[30].
The prognostic value of sST2 appears to be valid for patients with HFpEF as well as for those with HFrEF, the internationally recognized cut-off level for sST2 being established at 35 ng/mL[11][23]. This cut-off value is based on numerous studies concerning the outcome of patients with HF. For instance, baseline sST2 <35 ng/mL was associated with better prognosis, expressed as a longer time span until the occurrence of a cardiovascular event (HR 0.30, 95% CI 0.14 to 0.63, p = 0.002). Similarly, a switch of sST2 values from <35 to >35 ng/mL was suggestive for a shorter interval until a cardiovascular event occurred (HR 3.64, 95% CI 1.37 to 9.67, p = 0.009)[11].
In this context, cardiac biomarkers that assess inflammation-related modifications in collagen or other profibrotic molecules could be used for early detection of HFpEF and for an improved therapeutic approach. Thus, progressive changes during seriated sST2 assessments may detect the earliest transition from only structural, subclinical myocardial injury to HFpEF. Elevated sST2 levels reflect cardiac remodeling and progression of fibrogenesis in myocardial tissue, these alterations being associated with LV diastolic disfunction and other mechanical changes, particularly of the left atrium[25].
Even if the lower absolute values of sST2 in HFpEF compared to HFrEF may suggest only a mild fibrogenesis, the stronger association with poor outcome in patients with HFpEF actually highlights the major prognostic importance of this biomarker, both at first medical contact and in successive follow-ups[19]. Noteworthy, sST2 is associated not only with cardiovascular events or death, but is also a strong predictor for all-cause mortality or rehospitalization in patients with HF, irrespective of ejection fraction[20][23].
An interesting viewpoint is to perform seriated determinations in order to guide therapy adjustments. Recent results claim that the essential principle is to have a baseline sST2 value at admission, and then another one in the following four to six days, which will be decisive whether to initiate a more aggressive therapy according to the sST2 cut-off of 35 ng/mL[25].
Another concept focuses on a “high-risk” cut-off value of 70 ng/mL, which is allegedly a better option in distinguishing dyspneic patients with more severe acute HF. Moreover, this category of patients should be immediately hospitalized, with the prompt initiation of more aggressive anti-remodeling therapies in the context of a significant activation of the neurohormonal and fibrotic pathways. In Figure 1 there is a proposed algorithm for the management of patients presenting in emergency room with dyspnea or other acute HF manifestations (e.g., swollen ankles), depending on their sST2 levels[31].
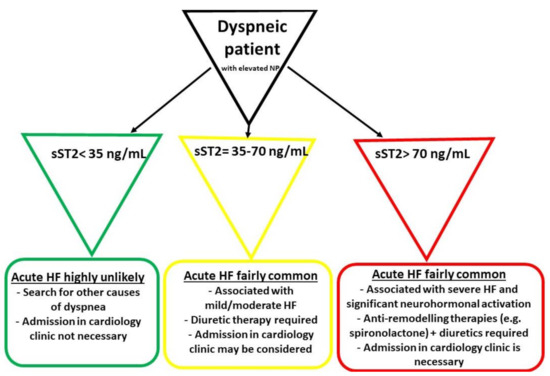
Figure 1. The initial management of patients presenting in emergency room with dyspnea and elevated natriuretic peptides (NP) (adapted from Aleksova et al.)[31]. HF—heart failure.





