1000/1000
Hot
Most Recent
 +1 point
+1 point
Perinatal infection seems to limit the neuroprotective efficacy of therapeutic hypothermia. Efforts made to use therapeutic hypothermia in LMICs treating NE has led to increased neonatal mortality rates. The heat shock and cold shock protein responses are essential for survival against a wide range of stressors during which organisms raise their core body temperature and temporarily subject themselves to thermal and cold stress in the face of infection. The characteristic increase and decrease in core body temperature activates and utilizes elements of the heat shock and cold shock response pathways to modify cytokine and chemokine gene expression, cellular signaling, and immune cell mobilization to sites of inflammation, infection, and injury. Hypothermia stimulates microglia to secret cold-inducible RNA-binding protein (CIRP), which triggers NF-κB, controlling multiple inflammatory pathways, including nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasomes and cyclooxygenase-2 (COX-2) signaling. Brain responses through changes in heat shock protein and cold shock protein transcription and gene-expression following fever range and hyperthermia may be new promising potential therapeutic targets.
Neonatal asphyxia describes a condition in newborns due to deprivation of blood carrying oxygen and nutrients (hypoxia-ischemia, HI) from the placenta to the fetus before or during delivery. Hypoxic-ischemic encephalopathy (HIE) is the feared neurological consequence that may occur in newborns following neonatal asphyxia causing brain inflammation and immunodepression [1]. Neonatal encephalopathy (NE) occurs in one to three of every 1000 births in high-income countries (HICs) [2][3] and approximately 10 to 20 of every 1000 births in low- and middle-income countries (LMICs) [4]. It is estimated that approximately 10% of the affected newborns die in their postnatal age, 25% develop severe and permanent neurological disabilities [5] such as cerebral palsy, seizures, mental retardation, learning impairment, and epilepsy [6][7][8][9]. Global statistics have shown that about 99% of annual neonatal deaths occur in the LMICs, and 1% in HICs [10].
For decades, multiple pre-clinical studies have been employed either using animal models of global or focal HI or cell culture models of oxygen-glucose deprivation, investigating the ameliorating effects of many chemical compounds on neuronal lesions. Recent pre-clinical and clinical research has shown that certain compounds have neuroprotective effects, suggesting that their use could be generalized for clinical practice in the near future. Additionally, the application of therapeutic hypothermia immediately after the hypoxic-ischemic event could prolong the window of opportunity for pharmacological therapeutic interventions. Therapeutic hypothermia is the standard treatment for NE of presumed HI origin in the HIC [11]. The controversy among physicians remains on whether hypothermia can also be administered safely and provide neuroprotection in other diseases, like traumatic brain injury or metabolic diseases. Therapeutic hypothermia has demonstrated efficacy in preventing perinatal brain injury following HIE [12]. There are several clinical trials and meta-analyses of newborns with HIE [13][14], showing neuroprotection from therapeutic hypothermia in HIC. However, it is not clear if therapeutic hypothermia is neuroprotective following birth asphyxia in LMIC. Emerging data have shown that therapeutic hypothermia is not safe for neonates in LMIC [15]. Also, the decrease in neonatal body temperature below 35 °C (accidental hypothermia) is the major cause of mortality in the LMIC. It has been shown experimentally that perinatal infection limits the neuroprotective effect of therapeutic hypothermia and clinically enhances neurotoxicity, and increases mortality in term asphyxiated neonates in LMIC.
Multiple etiological factors predisposing to NE have been described. They include antenatal maternal factors, hypoxia-ischemia, placental pathologies, neonatal stroke/thrombophilia, genetics and epigenetics, metabolic disorders, and perinatal infection (Figure 1). Infections during pregnancy can increase the expression of cytokines, causing inflammation to the fetal brain, leading to brain damage in the fetus and, subsequently, the newborn [16]. Some types of infection that have been linked with neonatal brain injury include viruses such as chickenpox, rubella, cytomegalovirus (CMV), and bacterial infections such as infections of the placenta or fetal membranes, or maternal pelvic infections.
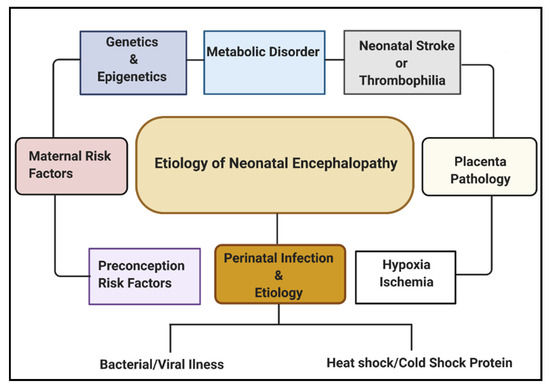
Regarding global neonatal mortality, a prospective study by Karsten et al. [17] reported that the global rate of severe infections accounted for 36% of all neonatal deaths, 29% were due to prematurity, and 23% were due to birth asphyxia. Evidence from recent studies has shown that HI brain damage plus perinatal intrauterine infection are the major etiological risk factors for cerebral palsy (CP). Both in vivo and in vitro neurologic assessments reveal that term infants born to mothers with clinical chorioamnionitis, suffered from a inflammatory cytokine storm, which is correlated with CNS abnormalities [18]. Also, several data have shown that both infection and HI in the brain promotes the production of proinflammatory cytokines, which may lead to further injury in the cerebral tissue [19][20][21].
Studies have shown that severe infection and prematurity-associated mortality is increased due to mild (33–36 °C) and moderate (28–32 °C) hypothermia [22]. Perinatal infection pre-sensitizes the fetal brain and makes it vulnerable to HI [23][24]. A previous study showed that therapeutic hypothermia was not neuroprotective in a LPS-sensitized HI brain injury model in newborn rats [25]. The authors observed a dramatic loss of brain area after inflammation-sensitized HI brain injury, which was not reduced by therapeutic hypothermia.
Perinatal infection stimulates the innate immune and inflammatory responses, causing the intracellular production of pro-interleukin 1 beta (IL-1β) following the stimulation of the pattern-recognition receptors (PRRs) such as the Toll-like receptors (TLRs) and leading to subsequent cell death [26]. The innate branch of the immune system relies heavily on TLRs and Nod-like receptors (NLRs) to detect and dissemble invading pathogens. Most of the important cell types expressing TLRs are the antigen-presenting cells (APCs), including macrophages, dendritic cells, and B lymphocytes [27]. Experimentally, TLRs are identified in most cell types and can be expressed either constitutively or inducible during infection [28][29][30]. The expression of TLRs and NLRs on and in both migrating and non-migrating cells is crucial for the rapid response to foreign invaders. The severity of a perinatal infection-induced inflammatory storm in neonates is strongly associated with nuclear factor kappa B (NF-κB) activation. It leads to nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation, followed by an increase of IL-1β expression, up-regulation of cyclooxygenase-2 (COX-2), and transient receptor potentials (TRPs). The up-regulation of COX-2 and TRPs acting as both sensor and effector shuffling among the nervous, vascular, and immune system will be discussed later. The activation of functional pro-IL-1β requires proteolytic cleavage, predominantly by caspase-1, and a component of the NLRP3 inflammasome multi-protein complex, resulting in secretion of mature biologically active IL-1β (Figure 2) [17]. Activation of the NLRP3 inflammasome triggers local mediators of the host cell damage in vivo, such as free radicals and DNA or adenosine triphosphate (ATP).
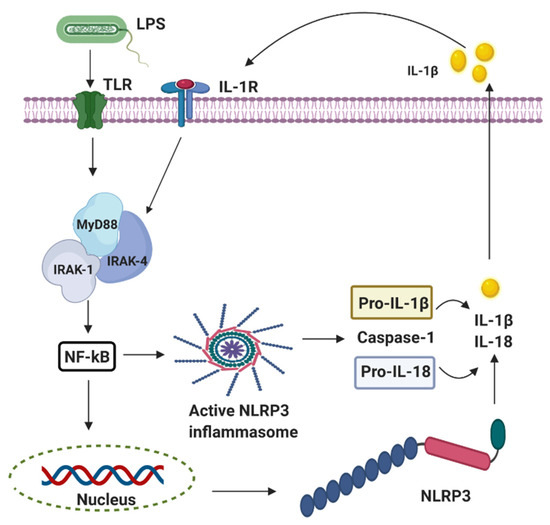
Figure 2. The activation of nod-like receptor (NLR) family pyrin domain containing 3 (NLRP3) inflammasomes involves multiple endogenous or exogenous stimuli. LPS stimulation stimulates Toll-like receptors (TLRs), which lead to the up-regulation of NLRP3, IL-1 via nuclear factor kappa B (NF-κB) dependant Myeloid differentiation factor 88 (MyD88), IL-1 receptor-associated kinase 1/4 (IRAK4).
COX-2 is an eicosanoid that generates arachidonic acid-derived lipid autacoids, including prostaglandins (PGs), thromboxanes, and leukotrienes. COX-2 is a critical mediator of inflammation, resolution, and tissue homeostasis. It is involved in a broad range of physiological processes, such as inflammation, fever, allergy, and pain. The presence of infection triggers the upregulation of inositol-requiring enzyme 1α–X-box binding protein 1, a branch of the endoplasmic reticulum (ER) stress pathway, to direct the expression of microsomal prostaglandin E synthase-1 and prostaglandin-endoperoxide synthase 2 (COX-2), which mediate the biosynthesis of prostaglandins (PGE2, PGD2, and PGF2α) from arachidonic acid. COX-2 production is associated with the production of a cytokine storm. Accordingly, ER stress signaling alone can cause a slight increase in the level of IL-6 (Figure 3). All together, in the presence of PGE2, interferon-γ, and activated endoplasmic reticulum stress, IL-6 production is greatly enhanced in glial cells. However, the key role of COX-2 in neonatal asphyxia inflammation and resolution has not been fully understood; studies have shown that cytokine overload in neonatal asphyxia contributes largely to morbidity and mortality [31]. As increased proinflammatory cytokines remain the driving force in severe neonatal asphyxia, the up-regulation activity of COX-2 following neonatal asphyxia may regulate the cytokine storm through fever induction. Fever is one of the usual clinical features that appear during the course of several infectious diseases. Fever is a process in which the body temperature rises, deviating from normal values, and according to Saladin and Porth [32], fever is a beneficial process as long as it does not persist or reaches 44 °C to 46 °C, where it could be fatal or lead to irreversible brain damage. Fever has been demonstrated to affect other immune cells as reflected by Harden et al. [33][34], including different types of innate immune cells such as neutrophils, monocytes, and T-cells or Natural Killer cells (NK) [35].
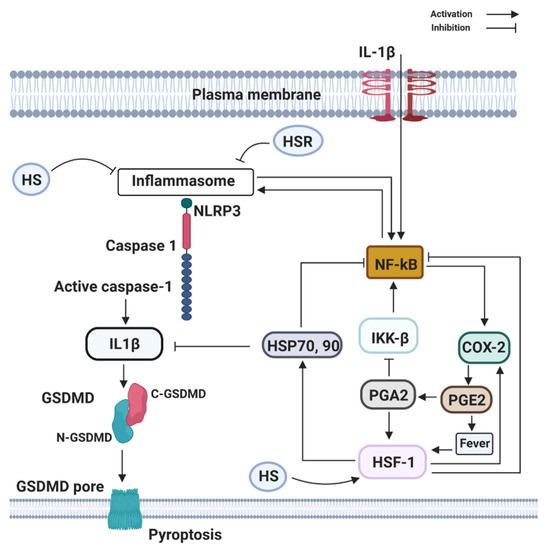
Figure 3. Components of the heat shock response (HSR) (HSP70 and HSF1) can directly or indirectly block the activation and transcribing activity of NF-κB. HSR is mainly centered on the heat shock transcription factor-1 (HSF1) that leads to the large production of the 70 kDa family of heat shock proteins (HSP70, 90). HSF1 may be directly activated by PGE2-induced rise in temperature (fever), by heat shock (HS), by estrogen (E2), and PGE2-derivative PGA2, whose physiological production is enhanced at late stages of inflammation. Activation of NLRP3, recruit the inflammasome adaptor ASC, which engages caspase-1. Subsequently, caspase-1 cleaves precursor IL-1β and IL-18 to their bioactive fragments, and also Gasdermin D (GSDMD) to trigger GSDMD N-domain pore formation in the plasma membrane. The GSDMD pores allow efficient IL-1β, eventually, cause the lytic cell death known as pyroptosis.
Fever has been suggested to be an essential product of several biological processes, where the detection of unchained pathogens sets up events that end up in favor of the host [36]. It is of paramount importance to understand the mechanisms of infection, where potential effects of fever on this process may have been overlooked. The induction of fever during neonatal asphyxia is associated with COX-2 expression, and the exposure of humans and rodents to temperatures ranged between 41–43 °C can induce heat shock response (HSR), leading to induction of heat shock protein (HSP) synthesis [37]. HSR is connected with immune responses and attenuates cytokines release [37], important for the host immune response and pathogen mechanisms of evasion. Pathogen-induced overexpression of HSPs is fundamental for the survival of the host organism during macrophage infection [38][39][40].
Heat shock proteins (HSPs) are present in all organisms and cell types. They are phylogenetically conserved proteins having both structural and functional significance, and they can be stimulated by stress signals (e.g., heat shock) and pathophysiological states (e.g., fever, inflammation, and infection) as well as those induced by normal development stress [41][42][43]. It has been shown that increased core body temperature has a protective role in the outcome of infection. Ostberg et al. [44] and others Jiang et al. [45] showed that mild systemic heating at 39.5 °C enhances the concentration of tumor necrosis factor alpha (TNF-α) and IL-6 in the blood and tissues of mice sensitized with bacteria endotoxin LPS. Heat shock proteins’ function in protein folding prevents protein denaturation or cell death under stressful conditions [46][47]. Although HSPs are intracellular proteins, they can be recruited to the plasma membrane or released into the extracellular environment and have immunomodulatory functions [48]. Most HSPs are involved in the synthesis and release of proteins from various cells either during cell injury or during translocation to the plasma membrane and are then secreted [49].
HSPs are known to have both positive and negative effects in regulating macrophage function, and this may depend on the cellular location of these HSPs. It is proposed that extracellular HSPs might serve as a danger signal to the immune response, whereas intracellular HSPs could serve as a negative regulator to control the inflammation [50]. Previous studies have shown that extracellular HSPs exert immune-stimulatory effects [48]. Wang et al. [51] demonstrated that extracellular HSP70 binds to lipid raft microdomains on the plasma membrane of macrophages and enhances their phagocytic ability. In fact, HSP70-mediated phagocytosis is very crucial for the internalization of antigens to CD4+ T-cells. It is clear that extracellular HSPs can largely stimulate the release of TNF-α, IL-6, IL-1β, IL-12, and nitrous oxide (NO). As well, chemokines are released by monocytes/macrophages [52][53][54][55], orchestrated through the CD14/TLR (both TLR2 and TLR4) complexes-activating downstream NF-κB and mitogen-activated protein kinase (MAPK) pathway [56][57][58]. In addition, HSPs also assists in the trafficking and targeting complex toward the Golgi apparatus [59]. This indicates that the elevation of extracellular HSPs may serve as endogenous danger signals to alert the host defense system through their cytokine-like function. HSPs have been studied for their potential to protect the brain from ischemic injury. They protect from both global and focal ischemia in vivo, and cell culture models of ischemia/reperfusion injury in vitro [60]; however, the mechanism of protection is not well understood. Although several members of the HSPs have shown to function as anti-apoptosis after HI brain injury, overexpression of HSP70 prevents the release of cytochrome c from mitochondria and the activation of casepase-9 by binding to apoptotic protease activating factor 1 (Apaf-1), thus blocking the caspase-dependent apoptotic pathway [61]. HSP90 binds to phosphorylated protein kinase B (Akt/PKB) and promotes the phosphorylation of the pro-apoptotic proteins Bax and caspase-9, and blocks the mitochondrial apoptosis pathway [61].
One member of the HSP90 family is glucose-regulated protein 94 (GRP94). Its expression has been shown to inhibit the activation of caspase-3 and calpain, maintaining the intracellular calcium homeostasis to protect neurons [61].
Transient receptor potential vanilloid channel 1 (TRPV1) is highly expressed in high temperate nerve fibers and is activated by heat, protons, and both endogenous and exogenous agonists [62][63]. TRPV1 is a nonselective cation channel that plays a significant role in thermoregulation, although the exact mechanisms in thermal regulation have yet to be fully understood [62][64][65]. Intravenous infusion of dihydrocapsaicin (DHC), a chili-derived TRPV1 agonist, and other capsaicinoids have shown the capacity to produce hypothermia in rodents and larger mammals [66]. In contrast, perinatal infection-induced hyperthermia was shown to occur through the TRPV1 channel [67][68][69][70]. It appeared that TRPV1 antagonists cause hyperthermia by blocking the tonic suppression of the autonomic cold defenses: thermogenesis and skin vasoconstriction [67][71]. In the brain, TRPV1 mediates cellular processes such as synaptic transmission, neurogenesis, and neuroinflammation [72][73]. Recently, TRPV1 has gained attraction from its functional expression in microglia and astrocytes [74][75]. It stimulates janus kinase 2- signal transducer and activator of transcription 3 (JAK2-STAT3) to regulate astrocyte and microglial activation and the expression of IL-1β and IL-6 [76][77][78][79]. TRPV1 deficiency in microglia and astrocytes has been shown to attenuate the expression of ionized calcium-binding adapter molecule 1 (Iba1), glial fibrillary acidic protein (GFAP), and IL-1β by reducing phosphorylation of NF-κB, JAK2, and STAT3. As well, a decrease in IL-1β is associated with TRPV1 deficiency by inhibiting activation of NLRP3 inflammasome. Not much is known about the role of TRPV1 in perinatal infection and birth asphyxia. However, in a selective study of the TRPV1 receptor, it was evident that TRPV1 expression is associated with a protective effect in the onset of sepsis after endotoxin [80]. Additionally, neonatal HI-induced neuro-behavioral disorders were significantly improved in mice lacking TRPV1 [81].





