1000/1000
Hot
Most Recent
 +1 point
+1 point
Chromosomal rearrangements comprise unbalanced structural variations resulting in gain or loss of DNA copy numbers, as well as balanced events including translocation and inversion that are copy number neutral, both of which contribute to phenotypic evolution in organisms. The exquisite genetic assay and gene editing tools available for the model organism Saccharomyces cerevisiae facilitate deep exploration of the mechanisms underlying chromosomal rearrangements.
Chromosomal rearrangements, including deletions, duplications, translocations, inversions, and formation of extrachromosomal circular DNA (eccDNA), are ubiquitous in cancer genomes and multiple genetic diseases. It was found that the translocation between chromosome 6 and chromosome 4, leading to the fusion of proto-oncogene 1, receptor tyrosine kinase (ROS1) to solute carrier family 34 member 2 (SLC34A2), drives lung cancer development [1], and the translocation-mediated fusion of the nucleoporin 98 kDa (NUP98) gene and topoisomerase (DNA) IIB 180 kDa (TOP2B) gene acts as a pathogenic factor in acute myeloid leukemia [2]. Recent studies have reported that eccDNA with amplified oncogenes is widespread in cancers [3][4]. The enhanced expression levels of oncogenes in eccDNA-carrying cells can result from eccDNA-mediated copy number changes as well as a higher chromatin accessibility of eccDNA due to the absence of higher-order compaction [5][6][7]. In addition to being a driver of carcinogenesis, chromosomal rearrangement also underlies the phenotypic diversification and environmental adaptability in organisms [8][9][10][11]. Multiple experimental systems have been developed in model organisms to decipher the rates, qualitative spectra, genetic dependencies, and phenotypic effects of chromosomal rearrangements.
The integrity of genomic DNA of cells is constantly challenged by both endogenous and exogenous agents. Of the many different classes of DNA lesions, double-stranded breaks (DSBs) are most deleterious and can be lethal to cells if left unrepaired [12]. Nonhomologous end joining (NHEJ) and homologous recombination (HR) are two evolutionally conserved pathways for DSB repair. Below we will briefly summarize how DSBs are processed and sealed, and discuss how this process results in chromosomal rearrangements in S. cerevisiae.
In S. cerevisiae, the trimeric complex Mre11-Rad50-Xrs2 (MRX) recognizes DSBs and initiates end resection. It removes oligonucleotides from the 5′ end when Sae2 is recruited, resulting in a limited length of single strand DNA (ssDNA) tracts (50–100 nt) [13][14][15]. Mre11 is a member of the lambda phosphatase family, exhibiting an ssDNA endonuclease activity and a 3′ to 5′ dsDNA exonuclease activity that processes DSB ends. MRX also recruits 5′ to 3′ exonuclease Exo1 or ssDNA endonuclease Dna2 to enable extensive resection (thousands of nucleotides or even longer) in concert with the RecQ family DNA helicase Sgs1 [15][16]. What is the purpose of extensive resection? Firstly, extensive resection is critical for template searching during homology-dependent repair [13][14][16][17]. Secondly, extensive resection ensures fidelity by preventing repair between short dispersed repeats [16][17]. Thirdly, long ssDNA tails are required for cells to signal DSBs and activate DNA damage checkpoints [17]. Nevertheless, it should be noted that long 3′ ssDNA tracts facilitate multiple template invasion and complex chromosome rearrangements [18][19].
NHEJ, which does not require 3′ ssDNA overhang, takes place throughout the cell cycle. In the canonical NHEJ (c-NHEJ) pathway, the heterodimer Ku70/Ku80 recognizes and binds the broken ends to prevent resection [20][21]. The DNA ends can be ligated directly by ligase IV (Dnl4) after simple end trimming (up to 4 nt) [22]. In addition to c-NHEJ, alternative end joining (alt-NHEJ) and single-strand annealing (SSA) have been identified as backup NHEJ. Alt-NHEJ, also known as micro-homology-mediated end joining (MMEJ), is initiated by limited end resection (5 to 25 nt) [23]. The exposed 3′ ssDNA of the two broken ends are subsequently annealed, followed by single-stranded gap filling and ligation. Extensive resection resulting in long stretches (>30 nt) of 3′ ssDNA is suitable for single-strand annealing (SSA) [24]. The long 3′ ssDNA tails are first bound by replication protein A (RPA) to prevent secondary structure formation, followed by Rad52-mediated annealing. Distinct from NHEJ, HR takes place in S and G2 phases in the presence of sister chromatids. As mentioned above, extensive resection is a prerequisite for homolog search and alignment. In HR, the long 3′ ssDNA coated by Rad51 searches for a homologous template to invade, forming a D-Loop [25]. Detailed information about how the extent of DNA end resection and other factors that determine the choice of repair pathway (including the nature of the DSB ends, cell cycle stage, and multiple repair proteins) has been discussed in other recent reviews [26][27][28][29][30].
While c-NHEJ is a rapid and efficient way to repair DSBs, it is usually regarded as an error-prone repair pathway because it rejoins the DSB ends without the use of an intact template (Figure 1). C-NHEJ often leads to small deletions and insertions that can be easily verified and selected by frameshift reversion assays in S. cerevisiae [31]. C-NHEJ also readily causes large deletions and translocation [32], respectively, when two DSBs occur simultaneously at the same and different chromosomes [33][34]. Cells in which c-NHEJ is blocked are still able to repair DSBs through alt-NHEJ, which is a more error-prone pathway compared to c-NHEJ. As shown in Figure 1, alt-NHEJ inevitably results in the deletion of inter-microhomology sequences and one copy of microhomology. Interestingly, polymerase θ associated insertions were occasionally observed at the DSB sites sealed by alt-NHEJ [35][36]. Chromosomal translocations via alt-NHEJ also elevate mutagenesis of the flanking of breakpoint junctions, suggesting that alt-NHEJ could destabilize genomes by triggering chromosomal arrangements and increasing the mutation rate [37]. In contrast to alt-NHEJ, SSA requires more extensive resection that results in large deletions (Figure 1) [38]. Long repeat sequences, such as Ty retrotransposon-elements, were proposed as important mediators of SSA that caused translocation in yeast [39][40]. Ramakrishnan et al. [41] reported that inverted DNA repeats placed near a DSB can generate a dicentric chromosome or a fold-back (hairpin) structure through SSA, indicating the orientation of repeats would influence the choice and outcome of DSB repair [42].
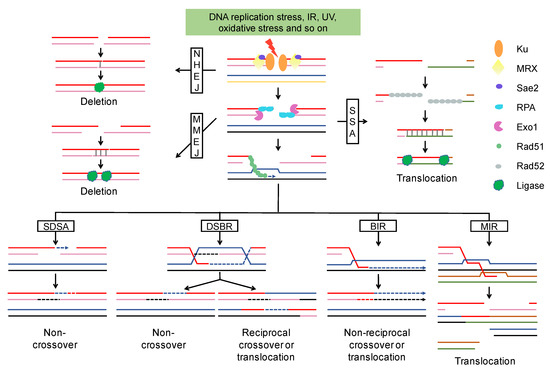
Figure 1. DSB end resection and repair pathway. After DSB formation, Ku binds to the DSBs and promotes the classic NHEJ pathway. NHEJ generally cause small deletions. The MRX-Sae2 complex initiates the resection and Exo1 extends the resection to expose longer 3′ ssDNA that is bound by RPA to prevent degradation. MMEJ and SSA are two alternative end-joining pathways, both of them can result in deletion or translocation. In the homologous recombination pathway, the 3′ ssDNA is coated with Rad51 and invades the homologous template to form a D-Loop. DNA synthesis is represented by an arrow and newly synthesized DNA by a broken line. Homologous recombination leads to multiple outcomes (non-crossover, crossover, gene conversion, and translocation) depending on what templates were used and in which way the D-Loop was processed. Abbreviations: NHEJ, Nonhomologous end joining; MRX, Mre11-Rad50-Xrs2 complex; RPA, replication protein A; MMEJ, microhomology-mediated end joining; SSA, single strand annealing; SDSA, synthesis-dependent strand annealing; DSBR, double-strand break repair; BIR, break-induced replication; MIR, multi-invasion-induced rearrangement.
HR starts with D-Loop formation and then proceeds via one of the following sub-pathways (Figure 1): (I) Synthesis-dependent strand annealing (SDSA). The 3′ end of the invading strand is used as a primer to extend the D-Loop through DNA synthesis. The newly copied strand is re-annealed to the other broken end to allow gap filling by a second round of DNA synthesis. (II) Double-strand break repair (DSBR). The D-Loop is stabilized via “capturing” of the second broken DSB end. The resulting double Holliday junction (dHJ) can be resolved by topoisomerase-mediated dissolution or by cleavage of the Holliday junctions. (III) Break-induced replication (BIR). The invasion results in the initiation of DNA synthesis that proceeds via a migrating bubble with asynchronous DNA synthesis distal to the point of invasion. (IV) Multi-invasion-induced rearrangement (MIR). The 3′ end invades two intact donors and stimulates translocation between the two donors without any homology.
HR, during which the broken chromosome is repaired using an intact sister chromatid or homolog as a template, is much more important than NHEJ in diploid cells of S. cerevisiae [43]. HR between homologous chromosomes leads to loss of heterozygosity (LOH), a common genomic alteration in cancers and diploid S. cerevisiae strains. SDSA and DSBR are the main pathways responsible for interstitial and terminal LOH, respectively [44]. DSBR would lead to reciprocal translocation if non-allelic homology was used as the template for DSBs repair [45]. While BIR plays a crucial role in replication fork restart and telomere maintenance due to its capability to repair one-end breaks, this pathway results in terminal LOH and translocation as well [46][47]. Additionally, half-crossovers (HC) are generated when BIR is interrupted and the migrating bubble is resolved, resulting in the fusion of donor and recipient chromosomes [47]. The remaining DSB end can invade another donor to initiate a new HC or other chromosomal rearrangements [47][48]. MIR is a newly characterized pathway that drives complex rearrangements [19]. As shown in Figure 1, MIR not only stimulates translocation, but also generates new DSBs that give rise to further chromosomal rearrangements [19][45][49][50].
In summary, although NHEJ and HR are critical for maintaining the integrity of genomic DNA, aberrant DSBs repair performed by these two pathways leads to diverse chromosomal rearrangements.
In wild-type yeast cells, the rate of chromosomal rearrangements during vegetative growth is low [43]. Most information about the rates of these events was based on the genetic assay system involving single genes or partial chromosomes [51][52]. More recently, subculturing of yeast strains over many generations for mutation accumulation (MA) followed by whole-genome sequencing has allowed a more global analysis of chromosomal rearrangements [53][54][55][56]. Sui et al. [53] performed MA experiments in a diploid S. cerevisiae strain (spo11/spo11; unable to enter meiosis), identifying chromosomal rearrangements throughout the genome by deep-coverage genome sequencing. This study detected 47 chromosomal rearrangements including 35 deletions, 12 duplications, and 1 translocation. It was calculated that spontaneous chromosomal rearrangements occur at a rate of 1.8 × 10−4 per cell division. This rate is one magnitude lower than that of LOH events (4.6 × 10−3 per cell division; gene conversion and crossover) resulted from SDSA and DSBR pathways (Figure 1) [53]. They also showed certain regions are more susceptible to rearrangements than other regions. For example, the region of 184–195 Kb (including three genes RRN11, CAT2, and VPS71) located between two flanking Ty elements (standing for transposons of yeast, which are dispersed repeat retrotransposons in the yeast genome) on chromosome XIII, and the region of the RDL1/2 locus on chromosome XV are two hotspots for interstitial deletion or duplication [53]. Non-allelic recombination between two sister chromatids by the DSBR pathway might be responsible for those frequent interstitial deletions and duplications (Figure 1). SSA to repair a DSB located between two direct repeats can be an alternative mechanism underlying spontaneous interstitial deletion (Figure 1). In one subcultured isolate, they also found recombination between two directly oriented Ty transposons on chromosome III that resulted in a circular DNA molecule [53]. Most strikingly, this work demonstrated that spontaneous chromosomal rearrangements generally involve homologous recombination between non-allelic dispersed repeats in the yeast genome. In a recent study by Sampaio et al. [55], the authors showed that two LOH events coincided at two different chromosomes at rates 14- to 150-fold higher than predicted if these two events originated independently of each other. This finding suggested that multiple genomic alterations can occur simultaneously during a limited time window, possibly as short as a single cycle of cell division [55]. What is the nature of recombinogenic lesions under spontaneous conditions? The study of St. Charles and Petes indicated that most mitotic recombination events were caused by DSBs in G1 phase [57]. Since the frequency of recombination in S. cerevisiae grown under anaerobic conditions was significantly lower than that under aerobic conditions [58], oxidative stress may constitute an important factor responsible for spontaneous chromosomal rearrangements.
The fidelity of DNA replication ensures precise genetic information passage in living organisms. At least three DNA replication polymerases (α, δ, and ε) are required to complete genome replication in all eukaryotes [59][60]. Defects in DNA polymerases, the presence of DNA lesions or secondary structure, origin re-firing, as well as limited concentrations of intracellular deoxyribonucleotide, would interrupt the normal function of replication forks and lead to DNA replication stress (DRS) that has been recognized as a hallmark in cancers [61][62][63]. Using S. cerevisiae models in which the levels of DNA polymerases were reduced, several studies have explored how DRS stimulates DNA lesions and chromosomal rearrangements [64][65][66][67][68]. In these studies, the expression of the genes POL1 (encoding the catalytic subunit of polymerase α) or POL3 (encoding the catalytic subunit of polymerase δ) was regulated by the galactose-inducible GAL1 promoter. When grown in low-galactose medium (0.005% galactose), the expression levels of the DNA polymerases were reduced which greatly elevated rates of genomic alterations. Using the whole-genome SNP microarray, Zheng et al. [65] detected the chromosomal rearrangements among 35 colonies derived from a low polymerase δ strain. Of the 41 interstitial deletions/duplications, 27 were within the tandem clusters (CUP1 and HXT6/7 genes), 4 were between solo long terminal repeats (LTRs), and 10 involved nonallelic Ty transposons. They also identified repeats that mediated 16 terminal alterations and most of them were paired events: a terminal deletion and a terminal duplication deletion occurred in the same strain. This pattern may be caused by a break on one chromosome that was repaired by BIR using an ectopic allele on a nonhomologous chromosome as a template (Figure 1). This work showed that DRS imposed by low levels of polymerase δ stimulate the frequency of chromosomal rearrangement by two orders of magnitudes [65]; the main source of chromosomal rearrangements is HR between ectopic repeats rather than NHEJ under DNA replication stress. By a genetic assay, the authors also showed that most recombinogenic DNA lesions in the low polymerase δ cells occurred during S/G2 phase, presumably as a consequence of replication fork collapse under DNA replication stress.
The DSB repair process can be broadly divided into two stages: DSB signaling and repair. These two processes coordinate to guard the genome integrity in cells [69]. In S. cerevisiae, Mec1-Ddc2 (orthologues of human ATR-ATRIP) and Tel1 (orthologue of human ATM) are two sensor kinases to perceive DSBs [70]. Mec1-Ddc2 senses ssDNA through interaction with RPA [71], while Tel1 is recruited to DSBs and activated by the MRX complex [72]. It was found that the mec1 tel1 null mutant showed a great increase (~13000 fold) in chromosomal rearrangement, of which most are chromosome translocations [73][74]. Downstream gene (such as RAD53, SML1, MRC1, and TOF1) knockout increased the frequency of chromosomal rearrangement as well [75]. These studies suggest that the deficiency in DNA damage checkpoints drives chromosomal rearrangements.
Although HR is the major genetic mechanism underlying chromosomal rearrangements in S. cerevisiae, loss function of HR (rad52 mutant or rad51 rad59 double mutant) results in a higher rate of chromosomal rearrangements due to the error-prone feature of NHEJ (see review [76]). The genes involved in DSB resection, which channel DSBs into HR, are particularly important for maintaining genome stability. The deletion of either MRE11 or XRS2 increased the rate of chromosome rearrangement by more than 500-fold [77][78]. Large palindromic duplications occurred at a higher rate in the sae2 mre11 double mutant than in wild-type cells [18]. The sgs1 mutant increas the frequencies of deletions and translocations that contain extra regions of imperfect homology at the breakpoints [79]. A defect in EXO1 caused about a 14-fold increase in chromosomal rearrangements [80], and the exo1 sgs1 double mutant showed a dramatic increase in chromosomal rearrangement of about 450 times [81]. The HR intermediates can be disrupted by multiple pathways [82][83][84][85], among which the DNA mismatch repair pathway can unwind heteroduplex HR intermediates by recognizing mismatches by Msh2-Msh6 or Msh2-Msh3 [86][87]. Defects in the DNA mismatch repair lead to close rates of recombination mediated by identical sequences and by imperfectly matched sequences [86].
Overall, DNA repair deficiency would dramatically increase the frequency of chromosomal rearrangements. It should be noted that the overactivity of certain HR components also brings risks to genome. For example, the 3′ to 5′ exonuclease activity of Mre11 contributes to large deletions and translocations due to the excessive process of DSBs [88], emphasizing that strict regulation of the DNA repair activity is pivotal to genome stability.
The integrity of genomic DNA is also challenged by exogenous factors that inflict damage upon DNA and induce chromosomal rearrangements [89][90][91]. Although IR is not a major force of spontaneous genomic alterations, it is extensively used as an exogenous factor to generate DNA damage and recombination in research. In the study of Argueso et al. [40], it was found that 7–28% of the G2-phase arrested yeast cells still survived after extensive irradiation that introduced about 250 DSBs per cell, demonstrating the ability of a eukaryotic genome to undergo extensive repair and rebuilding from extreme DSBs. Using pulsed-field gel electrophoresis (PFGE) analysis, they found nearly two-thirds of the appeared colonies (54 of 71) contained at least one chromosomal rearrangement. Comparative genomic hybridization arrays (CGHarrays) of IR-treated isolates revealed that almost all of the chromosomal rearrangements resulted from HR between nonallelic repetitive elements (mainly Ty retrotransposons). These findings argued that HR is the primary pathway to repair DSBs in diploid yeast cells resulted from IR, and after IR treatment, chromosomal rearrangements generated by recombination between nonallelic repeats can profoundly reshape the genomes [40]. Interestingly, although ultraviolet (UV) is highly recombinogenic, very few chromosomal rearrangements were observed in UV-treated yeast cells [92]. This difference is likely to reflect the different numbers of DSBs caused by IR and UV. IR is able to cleave DNA directly, causing much more DSBs than UV [92].
Bleomycin (BLM) is a radiomimetic chemical used for the treatment of a variety of tumors [93]. Freeman and Hoffmann found the frequency of mitotic recombination in yeast could be significantly elevated by BLM treatment [94]. Using a whole-genome SNP array, Sheng et al. [95] detected 78 LOH events and 3 aneuploidy events among the 13 isolates derived from a heterozygous diploid S. cerevisiae strain QSS4 after treatment with 4 μg/mL Zeocin (a BLM analog). The authors also detected an interstitial deletion mediated by an LTR sequence [95]. This study demonstrated that a DNA damaging chemical can greatly stimulate mitotic recombination that leads to LOH and chromosomal rearrangement in yeast [95]. The ratio of chromosomal rearrangement and the LOH event was similar to that observed in the wild type cells. Etoposide is a podophyllotoxin derivative that belongs to the class of topoisomerase poisons. Exposure to etoposide leads to ssDNA breaks by trapping the topoisomerase 2 (Top2)-DNA complex, which can be converted to DSBs after chromosomal replication [96]. It has been demonstrated that Top2 poisons can trigger chromosomal translocations and shorten the replicative lifespan of yeast [97][98]. Camptothecin (CPT) also indirectly causes DSBs by stabilizing the covalent Top1-DNA cleavage intermediate [99]. Treatment with 50 μg/mL CPT increased the frequency of chromosomal rearrangements by about 50-fold in yeast [51]. Further, copy number variations of rDNA and CUP1 clusters were observed after CPT treatment [100]. Methyl benzimidazole-2-yl-carbamate (MBC or carbendazim) is a widely used broad-spectrum benzimidazole fungicide that interferes with the function of the mitotic spindle, resulting in whole chromosomal aberration (monosomy, trisomy, and uniparental disomy). Additionally, it was also reported that MBC treatment led to chromosomal rearrangements [101][102]. Because of its potential to stimulate large-scale genomic alterations, MBC has been used as a mutagen to obtain S. cerevisiae mutants with improved stress tolerance and fermentation performances in several studies [103][104]. The primary conclusion from the above discussion is that any chemical that interrupts DNA replication or separation may be a potent inducer of chromosomal rearrangements.
Oxidative stress is a constant threat to the genome stability of aerobic organisms [105]. Reactive oxidative species (ROS)-induced DNA damage includes various general damages as well as DNA breaks [106]. In the study by Zhang et al. [58], yeast cells treated with 100 mM H2O2 for 1 h resulted in a one hundred-fold elevation of the frequency of mitotic recombination in a diploid S. cerevisiae strain, demonstrating that H2O2 is a potent inducer of genome instability. The authors observed 10 deletions and duplications, half were interstitial and half were terminal, among 30 isolates obtained after repeated exposure to H2O2. Similarly, chromosomal rearrangement can result from mutations of genes (such as SOD1 and TSA2) involved in the anti-oxidative systems [58][107] and intracellular ROS accumulation induced by extracellular stressors. Qi et al. [108] found that exposure to furfural, a major inhibitor existing in the cellulosic hydrolysate used for bioethanol fermentation, led to enhanced rates of chromosomal rearrangements in yeast cells. Although furfural cannot cleave DNA directly, the authors observed frequent in vivo DNA breaks due to accumulation of intracellular ROS [108]. It is likely that prolonged exposure of yeast cells to the recombinogenic agent under industrial conditions could quickly lead to loss of selected desirable traits of industrial yeast strains. These findings have direct implications for the use of yeast cells in the production of bioethanol from cellulosic feedstocks, particularly in distilleries that utilize cell recycling.





