1000/1000
Hot
Most Recent
 +1 point
+1 point
The process by which dysfunctional mitochondria are selectively targeted for lysosome-mediated degradation otherwise known as mitophagy, requires the serine/threonine kinase PINK1 and the E3 ubiquitin ligase Parkin to occur. In the last decade the PINK1/Parkin pathway received great attention due to its importance in many physiological and pathological processes. Understanding the mechanisms by which mitochondria are selectively recognized and targeted for degradation is thus fundamental to understand and to develop therapies for many devastating diseases. Here the mechanisms at the basis of the PINK1/Parkin-mediated degradation of dysfunctional mitochondria are described.
Macroautophagy (referred hereafter as autophagy) is a genetically programmed process that removes unnecessary or dysfunctional cellular components and recycles them[1]. In this process, firstly, a double-membrane structure, known as the autophagosome, is created to engulf targeted cellular elements and later fuses with lysosomes to form autolysosome, where the enveloped contents are degraded. More than 30 autophagy-related genes (Atg) have been identified in yeast, and most of them have mammalian homologues[2]. Initially, autophagy was defined as a non-selective pathway, but it is now widely recognized that there are two different types of autophagy—non-selective and selective. Under nutrient deprivation and during development, the non-selective pathway is mainly activated in order to provide nourishment for cell survival. The selective pathway, instead, is active even in nutrient-rich conditions and plays an important housekeeping function, as it is activated to eliminate dysfunctional organelles, protein aggregates, or intracellular pathogens[3]. Selective autophagy requires specific receptors able to recognize targeted ubiquitinated cargos and to recruit the autophagosome machinery[4]. The most characterized type of selective autophagy is mitophagy, which specifically targets damaged mitochondria to degradation[5]. It is a key cellular process, particularly important in post-mitotic and slow-dividing cells (such as neurons), as it promotes the turnover of mitochondria preventing accumulation of dysfunctional organelles.
Though the crucial role of defective autophagy in neurodegeneration is well established[6], its implication in PD has been particularly investigated. In this respect, Narendra et al.[7][8][9][10] firstly linked PINK1 and Parkin to mitophagy, shedding light into the PINK1/Parkin mitochondrial quality control pathway.
PINK1 is 581 amino acids long and contains an N-terminal mitochondrial targeting sequence (MTS), a transmembrane domain (TM), a highly conserved serine/threonine kinase domain, and a C-terminal auto-regulatory domain[11]. Under physiological condition, PINK1 levels are quite low because it is rapidly degraded. As a mitochondrial targeted protein, PINK1 is imported into mitochondria through the outer mitochondrial membrane (OMM)-localized TOM (translocase of the outer membrane) complex and the inner mitochondrial membrane (IMM)-localized Tim (translocase of the inner membrane) 23 complex[12]. Translocation of the positively charged MTS through the Tim23 complex is energetically driven by the electrical membrane potential (ΔΨm) across the IMM. After passing through the Tim23 translocase, the N-terminal MTS domain reaches the matrix, where it is cleaved off by the mitochondrial processing peptidase, MPPα/β. This pathway is called the “pre-sequence pathway” (Figure 1). Presenilin-associated rhomboid-like (PARL) is an IMM-resident protease that subsequently cleaves PINK1 in the TM domain between Ala103 and Phe104[13], producing a truncated 52 kDa protein (the full-length protein is 64 kDa) that is retro-translocated to the cytoplasm and degraded via the N-end rule proteasomal pathway[13]. There are still some controversial data on the precise PINK1 mitochondrial sub-localization—some evidence indicates that PARL may cleave PINK1 at the IMM while the PINK1 catalytic C-terminal domain remains in the cytosol. However, PINK1 has been implicated in the phosphorylation, not always clearly if in a direct or indirect manner, of different proteins that are resident inside mitochondria. Among them there are the High Temperature Requirement Protein A2, also known as HTRA2/Omi protease[14], the chaperone tumor necrosis factor receptor associated protein 1 (TRAP1)[15] in the IMS, and the complex I subunit NDUFA10 (NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 10) located in the IMM[16].
As mentioned before, mitochondrial import requires an electrochemical gradient across mitochondrial membrane, negative on the matrix side, and is inhibited by mitochondrial depolarized agents (e.g., Carbonylcyanure m-chlorophénylhydrazone, CCCP). When translocation within the Tim23 complex halts due to ΔΨm loss, full-length PINK1 is retained in the OMM, with the C-terminal facing the cytosol. Here, PINK1 forms a super-molecular complex (ca 700 kDa) composed of TOM complex subunits and dimeric PINK1 that facilitates its autophosphorylation on Ser228 and Ser402 residues in the kinase domain[17]. Recently, it has been suggested that the ADP/ATP translocase (ANT), which acts as proton gradient-dependent carrier in the IMM, acts as a bioenergetic sensor and is critically required for mitophagy[18]. Indeed, it seems essential for the suppression of TIM23-mediated protein translocation and subsequent stabilization of PINK1 upon mitochondrial depolarization, independently from its ADP/ATP exchange activity. Moreover, PINK1 has been the first identified mono- and poly-ubiquitin kinase[19][20] able to phosphorylate Ub at the conserved Ser65. Ubiquitination is a post-translational modification that typically marks proteins for degradation via proteasomal pathway[21]. However, it can also act as a signal for autophagy[22] and alters substrate activity and localization[23]. Ubiquitination, which is obtained through the covalent bond of ubiquitin to lysine residues or to the N-terminal of the amino group of substrate proteins, is carried out by the sequential action of three enzymes: E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin ligases. A substrate of the ubiquitination process can be also the ubiquitin (Ub) protein itself, as it contains seven lysine residues and an N-terminal, useful to build up polyubiquitin chain. The most common chain types are K48 and K63 chains, in which multiple ubiquitin molecules are linked in a linear arrangement with the C terminus of one molecule attached to lysine 48 (or 63) of the next. Parkin is a 465 amino acid protein of the RING-between-RING (RBR) family of E3 ubiquitin ligases, so-called because of the presence ring finger-type, or 'ring', domains separated by loops responsible for protein-protein interaction,[24][25] with lax substrate specificity[26][27]. It forms multiple types of ubiquitin chains, most frequently K63, K48, K11, and K6 linkages[28]. It is composed of a ubiquitin-like domain (Ubl) at the N-terminus, followed by four zinc-coordinating RING-like domains: RING0, RING1, IBR (in-between-RING fingers), and RING2[20]. RING domains bind E2 enzymes, though do not participate directly in the catalysis, functioning instead as a scaffold for the transfer of ubiquitin. Structural analysis showed that Parkin RING1 is the only domain similar to a classical RING finger motif, whereas the other three have a completely different structure, suggesting that RING1 is the E2 binding site on Parkin[29][30]. Differently, RING2 contains an active site cysteine (Cys431) that accepts ubiquitin via a thioester bond and then transfers it via an acyl-transfer to the substrate[8][31]. Parkin also contains two flexible linkers, one after the Ubl domain and the other between the IBR and the RING2 domains. The latter is the repressor element of Parkin (REP), named for its role in the regulation of Parkin activity. In normal conditions, Parkin is a cytosolic protein due to its structural auto-inhibitory mechanisms; indeed, the access to the catalytic RING2 domain is blocked by RING0 and, at the same time, the E2 binding site on RING1 is occupied by the Ubl domain and REP linker[32][33]. In addition to the regulation of cellular Parkin levels and activity, the Ubl domain is involved in substrate recognition, binding SH3 and ubiquitin interacting motif (UIM) domains, and associating with proteasomes[34][35][36][37][38]. Parkin itself becomes ubiquitinated by the attachment of K6 ubiquitin chains, which may play a role in its own degradation[28]. Ubiquitin Specific Peptidase 8 (USP8), a deubiquitinating enzyme (DUB), seems to be crucial for Parkin-mediated mitophagy—it preferentially cleaves K6-linked Ub chains from Parkin, a process required for the efficient recruitment of Parkin to depolarized mitochondria. As different studies have identify a role for other DUBs in Parkin-mediated mitophagy, it is becoming clear that, in addition to ubiquitination and phosphorylation, deubiquitination also plays a crucial role in this process[39].
PINK1 acts upstream of Parkin and is required for Parkin activation and recruitment to depolarized mitochondria[9]. In fact, it can phosphorylate Parkin Ubl domain at Ser65[40], inducing the loss of its auto-inhibitory conformation and the opening of its conformation. The binding of PINK1-phosphorylated Ub to the RING1 domain of Parkin facilitates Parkin phosphorylation by PINK1[41][42], inducing the further structural rearrangements and the activation of Parkin. Both phospho-Ub and phosphorylation of Ubl domain are required for Parkin full activation, with phospho-Ub serving also as a receptor for Parkin recruitment to mitochondria [43]. Taking into account all of these considerations, a positive feedback loop has been conceived to explain the PINK1/Parkin pathway in mitophagy[44]. The accumulation of PINK1 at the OMM leads to the phosphorylation of low basal levels of both ubiquitin and Parkin present on mitochondria, causing a positive effect on Parkin activity. Activated Parkin attaches Ub protein to OMM proteins, providing also more substrates to PINK1 phosphorylation, amplifying Parkin recruitment and activation. As result of this mechanism, dysfunctional mitochondria can be coated with phospho-ub chains. This amplifying positive feedback explains how high levels of Parkin can be recruited from the cytoplasm to the mitochondria membrane by low endogenous level of PINK1. Recent data also show the implication of the mitochondrial ubiquitin ligase MITOL/MARCH5, which belongs to the membrane-associated RING-CH E3 ubiquitin ligase (MARCH) family (also called MARCH5), one of the three mitochondria-localized ubiquitin ligases (E3s) identified thus far, in providing initial substrates to PINK1 [45]. It seems to act by controlling mitochondrial dynamics through the regulation of the mitochondrial fission/fusion factors such as Dynamin-1-like protein is a GTPase (Drp1) or Mitochondrial fission 1 protein (Fis1) and mitofusin 2 (Mfn2), respectively. In addition, it has been recently observed that a reduction of MITOL levels delays Parkin recruitment to depolarized mitochondria and decreases the efficiency of Parkin-mediated mitochondrial ubiquitination; conversely, its overexpression results in the opposite effect, suggesting that MITOL may introduce the initial “seed” for ubiquitination, promoting rapid Parkin recruitment at the onset of mitophagy (Figure 1).
The mechanism underlying the further degradation of Ub-primed mitochondria is still unclear, but it has been demonstrated that the PINK1/Parkin pathway is implicated also in the phase of the autophagy clearance of damaged mitochondria in several ways. Indeed, it promotes the fragmentation of mitochondrial network, allowing mitochondria to be taken up by autophagosome; it modulates the mitochondrial motility, in order to stop their movement; and it directly recruits the autophagic machinery to dysfunctional mitochondria[46]. PINK1 and Parkin, in fact, modify a wide range of substrate proteins in the OMM, mediating their clearance.
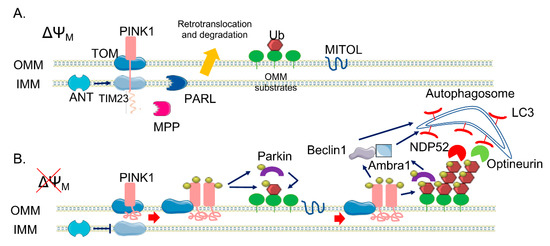
Figure 1. The canonical phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1)/Parkin pathway. (A) In healthy mitochondria, PINK1 is constitutively imported via translocase of the outer membrane (TOM)/translocase of the inner membrane (TIM)23 complexes to the inner membrane (IMM), cleaved by two proteases (mitochondrial processing peptidase (MPP) and presenilin-associated rhomboid-like (PARL)) and retro-translocated to the cytosol, where it is degraded. (B) When ΔΨM is dissipated, Adenine nucleotide translocator ANT inhibits TIM23-mediated import of PINK1, which is not processed and accumulates on the outer membrane (OMM). Here, a supercomplex is formed, composed by TOM complex subunits and PINK1 homodimers, facilitating PINK1 autophosphorylation and activation. Once activated, PINK1 phosphorylates ubiquitinated substrates on the OMM and thus recruits and phosphorylates Parkin. Phosphorylated Parkin starts to ubiquitylate several proteins on the OMM, which are new substrates of PINK1 phosphorylation—a positive feedback loop is initiated, leading to the coating of damaged mitochondria with phospho-ubiquitin chain, red arrows. The MITOL mitochondrial E3 ubiquitin ligase, seems to be fundamental for the introduction of the initial ubiquitination. Phospho-ubiquitin chains are bound by two mitophagy adaptors, Nuclear domain 10 protein 52 (NDP52) and Optineurin, blue arrows. The two adaptors recruit autophagosomes via Microtubule-associated protein 1A/1B-light chain 3 (LC3) binding, allowing the engulfment of dysfunctional mitochondria. In parallel, PINK1 interacts with Beclin-1 and Parkin with Ambra1, further stimulating autophagosome formation, blue arrows.
Interestingly, 36 OMM substrates of Parkin have been identified with high confidence[27], suggesting that no specific substrate is required for ubiquitin signalling of mitophagy. However, the best characterized targets are mitofusins 1 and 2 (Mfns 1/2)[47] and voltage-dependent-activated channel 1 (VDAC1)[10]. Mfns are involved in mitochondrial fusion and both Mfn2 and VDAC1 are involved in ER–mitochondrial tethering. As for Mfn2, the pool located on the ER membranes can form hetero- or homo-dimers in trans with mitochondrial mitofusins (i.e., Mfn1 or Mfn2) to fine tune the mitochondria-associated membrane (MAM)-dependent functions[48]. Although it is widely accepted that this protein is a major regulator of the mitochondria–ER interface, the exact role played at the mitochondria–ER contacts is still unclear. In fact, Mfn2 has been shown to have opposite effects in this interorganellar crosstalk[49][50], a fact that could be due to different distance ranges of contacts occurring at the ER–mitochondria interface[51]. As for VDAC1, it interacts with the ER Ca2+ channel inositol 1,4,5-trisphosphate receptor (IP3R) via the mitochondrial chaperone glucose-regulated protein 75 (Grp75)[52]. This complex seems to be essential for coupling Ca2+ transfer between ER and mitochondria[53]. Interestingly, very recently DJ-1 protein has been shown to take part of the VDAC1-IP3R_Grp75 complex and be essential for ER-mitochondria tethering[54].
The ubiquitination of both Mfns and VDAC1 and their subsequent proteasomal degradation facilitates the fragmentation of the mitochondrial network, and thus mitochondrial engulfment by autophagosomes. In addition, p97, an AAA+ ATPase, accumulates on mitochondria in a Parkin-dependent manner and promotes the degradation of OMM ubiquitylated proteins[55], leading to the rupture of OMM[56]. A recent study by McLelland and colleagues suggests that Parkin/PINK1 activation catalyses a rapid burst of Mfn2 phosphoubiquitination to trigger p97-dependent disassembly of Mfn2 complexes from the outer mitochondrial membrane, dissociating mitochondria from the ER. This promotes the availability of other Parkin substrates such as VDAC1, thus facilitating mitophagy[57]. Other substrates of the PINK1/Parkin pathway are Miro proteins (1/2), which are components of the primary motor/adaptor complex that anchor kinesin to the mitochondrial surface. Their degradation leads to the blockage of mitochondrial movement, promoting the segregation of dysfunctional mitochondria[58]. In addition, a recent paper by Safiulina et al.[59] suggested a role for Miro1 in Parkin recruitment to damaged mitochondria. In fact, in rat primary cortical neurons, Miro1 seems to be able to interact with Parkin also in the absence of PINK1 accumulation at the OMM and in basal conditions, suggesting a role for Miro1 as a mitochondrial docking site for the recruitment of Parkin from the cytosol. As mentioned above, other proteins have been identified as PINK1/Parkin substrates, but whether their ubiquitination could have a regulatory function rather than a degradative role remains unclear. Interestingly, recent studies have reported proteasome-independent-mediated ubiquitination by Parkin, which does not result in degradative ubiquitination but in fine tuning protein–protein association[60]. The PINK1/Parkin pathway not only primes damaged mitochondria by ubiquitination, but also promotes the induction of mitophagy. Two autophagy adaptors, namely, NDP52 and Optineurin[61][62] , are recruited by phospho-Ub to dysfunctional mitochondria, where in turn they recruit components of the autophagic pathway to initiate mitophagy. Moreover, Parkin interacts with Ambra1[63], a positive regulator of Beclin-1-dependent autophagy. Indeed, Ambra1 locally stimulates the activity of the class III Phosphoinositide 3-kinase (PI3K) complex, essential for the formation of new phagophores. PINK1 was also found to directly bind Beclin1 and to be required for the nucleation of the phagophores and the omegasome (the precursor of autophagosomes) generation[64]. The relative importance of mono- or polyubiquitinated chains on OMM substrates is still unknown, but monoubiquitin targeted to mitochondria is not sufficient to recruit Parkin, thus suggesting a possible specific role for polyubiquitin chains[65]. One last point to be mentioned is the role of phosphatase and tensin homolog (PTEN) itself, which seems to be directly involved in the modulation of mitophagy-related functions. Indeed a direct regulation of mitophagy through the promotion of Parkin recruitment to damaged mitochondria has been recently proposed for the first PTEN isoform identified so far, i.e., PTENα[66], whereas, interestingly, a newly identified long isoform of PTEN (PTEN-L) has been shown to act as a negative regulator of mitophagy by preventing Parkin mitochondrial translocation and inhibiting its E3 ligase activity[67]. Noteworthy, a fraction of PTEN has also been found to localize at the ER membrane and the MAMs and to regulate ER/mitochondria Ca2+ transfer[68].





