1000/1000
Hot
Most Recent
 +1 point
+1 point
Epidermal growth factor receptor (EGFR) amplification is a characteristic of the classical subtype of glioma.
With an overall survival of less than 35% in five years[1], malignant primary brain tumors are the most difficult to treat cancers. Of those, the most common type is represented by gliomas. Based on the expression patterns’ differences, glioblastomas are divided into three subtypes as follows: classical, proneural, and mesenchymal[2]. Because glioblastoma multiforme (GBM), a grade IV glioma[3], is one of the most aggressive primary brain tumors, recent studies and reviews have focused on deepening our understanding of the disease[4][5][6][7][8][9].
At present, GBM’s pathogenesis is better understood due to recent advances in molecular biology. For newly diagnosed glioblastoma, the current standard of care is represented by resection, followed by radiotherapy and temozolomide (TMZ) administration[10], but the median overall survival (OS) is not fully improved; therefore, new diagnosis and treatment strategies are needed[11][12].
Glioblastoma is the most common and the most deleterious glioma[13]. The 2011–2015 Statistical Report of the Central Brain Tumor Registry of the United States (CBTRUS) showed that glioblastoma represents 48% of the malignant brain and central nervous system tumors, with an incidence rate in the United States 1.58 times higher in males compared to females[14]. Due to the quick progression, even with aggressive multimodal treatment, glioblastoma remains almost incurable[15][16].
Nowadays, chemotherapy has a significant role in glioblastoma’s treatment strategies, with numerous research studies aimed to develop more efficient chemotherapeutic drugs[17]. Understanding the disease’s pathogenesis has a key role in identifying disease biomarkers and developing new potential chemotherapeutic drugs. We present some of the most promising signaling pathways involved in pathogenesis, with their specific targeting components.
GBM is characterized by nuclear atypia, cellular pleomorphism, mitotic activity, anaplasia, and rapid proliferation alternated with an aggressive invasion of the surrounding brain tissue. In its microenvironment, glioma cells are faced with many challenges such as acidity, hypoxia, and low nutrient availability. To maintain rapid growth, they need to modulate metabolic activity[18][19].
In multicellular organisms, tyrosine phosphorylation is involved in signal transduction, leading to differentiation, proliferation, migration, and survival[20][21].
Receptor tyrosine kinases (RTKs) are activated by binding their extracellular domain to corresponding ligands determining their oligomerization. This process activates the intracellular domain, facilitating the recruitment of proteins that start a signaling cascade, integrating numerous signaling pathways that lead to specific cellular responses[22]. Among all RTKs, epidermal growth factor receptor (EGFR) is the most amplified in GBM [23]. EGFR amplification is observed in the classical subtype of glioma[2]. EGFR gene amplification is detected in 57.4% of primary GBM patients, leading to high levels of EGFR protein, contributing to tumorigenesis and progression[24].
However, targeted therapies against this type of receptor have not yet shown a clear clinical benefit. Many factors contribute to resistance, such as ineffective blood–brain barrier penetration, heterogeneity, mutations, and compensatory signaling pathways.
The transmembrane receptor of tyrosine kinase epidermal growth factor (EGFR), also known as HER (human EGFR related) 1 or ErbB1, along with HER2/neu (ErbB-2), HER3 (ErbB-3), and HER4 (ErbB-4), is a member of the ErbB family and it is located on chromosome band 7p12 [25].
Like all RTKs, EGFR has an extracellular region, a single transmembrane domain, an intracellular juxtamembrane domain, a tyrosine kinase, and a C-terminal region. The ligands of ErbB receptors are divided into two main groups: EGFR activators called EGF agonists, and neuregulins that bind to ErbB3 and ErbB4[26][27].
The extracellular region of EGFR has two homologous domains (I and III) that bind ligands and two cysteine rich domains (II and IV)[28].
The juxtamembrane region tethers inactive EGFRs to the plasma membrane cytosolic surface, which contributes to EGFR activation[29]. Structural studies highlight the functional importance for certain regions, such as the structure of the first 30 amino acids from the intracellular juxtamembrane region of EGFR and the C-terminal 190 amino acids [27].
There are more than 40 EGFR ligands that control its signaling. They can be divided into high-affinity ligands, such as epithelial growth factor (EGF), heparin-binding EGF-like growth factor (HB-EGF), Transforming growth factor alpha (TGF-α), betacellulin (BTC), and low-affinity ligands, such as epiregulin (EREG), amphiregulin (AREG), and epigen (EPGN).
Expression of EGF-family proteins and activation of EGFR are features of cardiac disease[30][31]. Moreover, molecular alterations of EGFR include overexpression, deletion, or amplification, in different types of cancer. In GBM, EGFR amplification promotes invasion, proliferation, and drug resistance to radio- and chemotherapy[32].
Several trials on EGFR targeted therapy have failed to produce conclusive evidence, maybe because of the EGFR molecular heterogeneity in GBM, of the low specificity of the designed drugs, as well as because of low brain penetration[33]. Despite all this, the detection of EGFR alterations is still used as a prognostic marker for GBM because 24–67% of GBMs are characterized by a mutated gene, 40% by amplification, and 60% by EGFR overexpression[34].
In recent years, studies have proved that EGFR has pro-survival kinase-independent functions in malignant cells. This fact has offered a different perspective of understanding EGFR implications in cancer, with new ideas of EGFR targeted cancer therapy[35][36][37].
There are several different mechanisms of EGFR pathway activation, such as increased ligand production or overexpression/defective inactivation/mutation of the receptor. Many studies focused on the EGFR signaling mechanism in recent years, trying to conclude how the extracellular EGFR-ligand binding propagates through the single transmembrane helix (TM) to trigger intracellular kinase activation[38][39][40].
The expression of EGFR in normal cells is about 4 × 104–10 × 104 receptors/cell[41] whereas, in cancer cells, more than 106 receptors/cell are observed[42]
The EGFR RNA expression is increased by stimulating the EGFR-specific transcription factor (ETF). The receptor expression is regulated by epidermal growth factor (EGF) itself and other proteins such as E1A, Sp1, and AP2[36].
Like all RTKs, EGFR is activated by ligands featuring receptor-specificity. Briefly, ligand binding leads to a dimeric active conformation of EGFR by homodimerization (complexed with another EGFR) or heterodimerization (complex with another ErbB member). The tyrosine residues from other RTKs are autophosphorylated after ligand stimulation, and phenylalanine substitutions significantly impair the kinase signaling and the downstream signaling. Differently, EGFR Tyr-845 phosphorylation is not a required mechanism for ligand-induced EGFR activation, but it may represent the main mechanism for EGFR transactivation[43][44].
Proteins that express a proto-oncogene tyrosine-protein kinase (Src) homology domain 2 (SH2) region bind to the activated receptor, areactivated, and forward the signal to the downstream effectors, propagating critical cellular signaling pathways[45]. EGFR can simultaneously activate several signal transduction pathways such as phosphatidylinositide 3 kinase (PI3K) and serine–threonine kinase (AKT) and RAS/MAPK pathways[46].
For EGFR, the dimerization is completely receptor-mediated, with no physical interaction between two activating ligands. In normal physiologic status, the receptors are in a dynamic monomer–dimer equilibrium. In the absence of ligands, the extracellular domain presents a tethered configuration (intra-molecular links entirely block the dimerization arm), and the intracellular tyrosine kinase domain (TKD) is inactive. Ligand binding leads to a conformational change that exposes the buried dimerization arm, and the extracellular domain dimerizes, inducing conformational changes of the intracellular domain and enabling kinase activation[45].
A recent study by Chung et al. described physiological EGFR activation as being due to a ligand-mediated extracellular domain dimerization that stabilizes the N-terminal transmembrane dimer and disrupts autoinhibition, allowing the C-terminal juxtamembrane (JM-B) segment to stabilize the asymmetric kinase domain (KD) dimer, resulting in activation of EGFR signaling. They also concluded that the stimulus stabilizes the active KD conformation in pathological states and further the asymmetric KD dimerization. The inside–out coupling is weaker than the physiological outside–in coupling, suggesting that the extracellular (EC) dimer is linked through the N-terminal TM dimer with the asymmetric oncogenic KD dimer [38].
By ligand-induced dimerization, the cis-autoinhibition is released, and through a unique allosteric mechanism, the kinase activity of EGFR isactivated. It is well known that this mechanism consists of physical interaction between the C-terminal tail of the activator kinase and the other kinase N-terminal tail (receiver kinase) of the dimer pair, inducing conformational changes of the N-lobe of receiver kinase and trans-phosphorylation C-terminal tail of the activator [47].
EGFR activation and autophosphorylation result in the recruitment of downstream signaling proteins. Almost all autophosphorylation sites are binding sites for Src Homology 2-(SH2) or Phosphotyrosine binding-(PTB) signaling proteins. The SH2- proteins may be bound directly to the receptor, or indirectly through docking proteins using PTB domains[48]. EGFR can recruit and regulate many signaling pathways such as PI-3 K/AKT, RAS/MAPK, and JAK2/STAT. Therefore, EGFR functions as a hub involved in regulating various cellular processes[21][23], as shown in Figure 1.
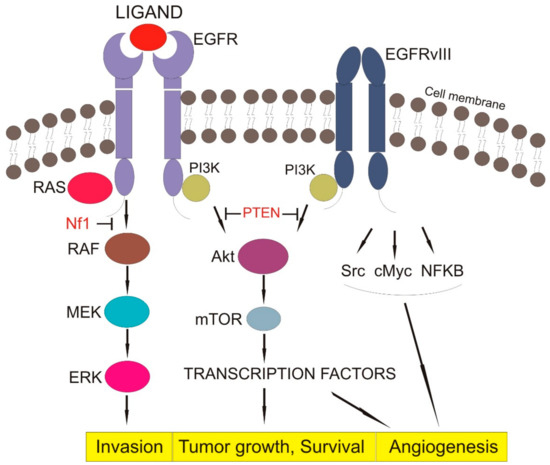
Figure 1. EGFR signaling pathway (EGFR—epithelial growth factor receptor, EGFRvIII—Epidermal growth factor receptor variant III, Pi3K—Phosphoinositide 3-kinase, RAS—family of genes involving cellular signal transduction, PTEN—Phosphatase and tensin homolog, NF1—Neurofibromatosis type 1, RAF—serine/threonine-specific protein kinases, MEK—Mitogen-activated protein kinase, ERK—extracellular signal-regulated kinase, AkT—Protein kinase B, mTOR—mammalian target of rapamycin, Src—Proto-oncogene tyrosine-protein kinase, cMyc—c proto-oncogene, NFKB—nuclear factor kappa-light-chain-enhancer of activated B cells, Block arrow—inhibition activity, Point arrow—pathway flow).
The PI-3K/AKT signaling pathway involves PI3K, an enzyme with SH2-signal transducer and its downstream effector AKT, regulating apoptosis and cell survival. Once EGFR is activated and phosphorylated, PI3K is brought to the cell membrane, and it phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2), forming phosphatidylinositol (3,4,5)-trisphosphate (PIP3). AKT reacts with PIP3, and it is phosphorylated at Threonin308 by phosphoinosite-dependent protein kinase-1 (PDK1) and at Serine 473 by the mammalian target of rapamycin complex 2 (mTORC2), reaching full activity. The phosphatase and tensin homolog (PTEN) negatively regulate the PI3K/AKT pathway by dephosphorylating and delocalizing PIP3 from the cellular membrane, resulting in the relocalization of AKT in the cytoplasm, where it is unable to be reactivated[49][50].
Class IA is one of the three different classes of PI3Ks featuring subunits with regulatory activity such as p85. Active EGFR achieves association with regulatory p85 through dimerization with human HER3, or via the docking protein GRB2-associated binder 1 (GAB1), relieving the inhibitory effect of p85[51]. GAB1 is a scaffolding protein involved in recruiting additional signaling proteins such as PI3K, SHP2, and p120RasGap. It is involved in many EGFR signaling outputs, and is the predominant mechanism linking EGFR to PI3K/Akt signaling[52][53].
Due to its increasing importance in different human cancers, GAB1 may represent an emerging potential therapeutic target.
The RAS/MAPK signaling pathway involves the growth-factor-receptor bound-2 (GRB2), which forms a complex with Son of Sevenless (SOS), a guanine-nucleotide exchange factor (GEF) and activates the RAS G-protein by exchanging guanosine diphosphate (GDP) with guanosine triphosphate (GTP)[54]. Consequently, RAS and mitogen-activated protein kinases (MAPKs) initiate a downstream signaling cascade to phosphorylate the nuclear protein Jun. Jun creates complexes with different nuclear proteins leading to the key transcription factor activator protein 1 (AP-1), responsible for translation and transcription of proteins involved in the growth and division of cells. Activated RAS is negatively regulated by GTPase activating proteins (GAPs), such as the tumor suppressor neurofibromin 1 (NF1) [55].
Signal transduction and activator of transcription 3 (STAT3) is tyrosine-phosphorylated or activated as pSTAT3 due to EGFR-regulation of interleukin-6 (IL-6) expression. This mechanism leads to a feed-forward in the IL-6/Janus kinase (JAK)/STAT3 loop [21][56][57][58].
The EGFR is one of the most frequently altered oncogenes in brain cancers. Except for hematopoietic cells, the majority of cell types express ErbB family members[35].
In glioblastoma cells, the EGFR tyrosine kinase activity may be dysregulated by multiple oncogenic mechanisms, such as gene mutation, overexpression of EGFR protein, increased gene copy number, rearrangements of chromosomes, and activation by autocrine function[59].
The EGFR gene is located on chromosome 7p11.2 and consists of 28 exons encoding a transmembrane protein receptor composed of 464 amino acids. Exons 5–7 and 13–16 encode the ligand binding domain, and exons 18–24 encode the tyrosine kinase domain. The region encoded by exons 25–28 is the site of autophosphorylation.
Although EGFR is one of the most important drug targets in cancer therapies, its mutations present an organ–site asymmetry, depending on the cancer’s organ of origin[60]. Although mutations occur in the kinase domain (KD) in other tumors, in gliomas, heterogeneous mutations and deletions are focused on the ligand-binding ectodomain (ECD). This tissue-specific feature leads to type-II tyrosine kinase inhibitors (TKIs) with high sensitivity for the inactive symmetric KD dimer (sKD), when administered in GBM mutations[61]. However, both intra- and extracellular GBM mutations result in ligand-independent oncogenic activation.
Almost 50% of the tumors characterized by EGFR amplification are positive for the mutant EGFRvIII and EGFR single nucleotide variants (SNVs). Due to this tumor-specific feature, novel therapeutic agents are currently under development to target the overexpressed EGFR or EGFRvIII proteins. An in-frame deletion of exons 2–7 characterizes the EGFRvIII, which results in overexpression of a truncated receptor that lacks some significant parts of the ECD. This prototypic oncoprotein is unable to bind ligands, and it is constitutively active. Several studies examined the effect of the EGFRvIII constitutive activity on the wtEGFR and ErbB2 protein levels. For example, one study evaluated the effect of Tyrphostin AG1478 on the protein levels and demonstrated that its administration increased protein levels of wtEGFR and erbB2 in vIIIA1 cells, due to the catalytic activity of EGFRvIII, while in its absence, the levels were reduced[62]. Furthermore, the unique peptide sequence of EGFRvIII generated by the fusion of exons 1 and 8 may serve as a tumor-specific target in immunotherapy[63], although subsequent phase III trial results are not as promising as initially anticipated[64].
A meta-analysis performed in 2017 by Felsberg et al. proved that EGFRvIII and EGFR SNVs do not represent prognostic keys in EGFR-amplified glioma patients. However, the amplification of EGFR is retained in recurrent glioma[63], although improved long-term survival by EGFRvIII therapy has been reported in glioblastoma patients [65].
Nevertheless, the research on EGFRvIII continues, producing inconclusive results. For example, Struve et al. just published in early 2020 the results of a study focused on the effect of EGFRvIII in regulating DNA mismatch repair. They tested if EGFRvIII influences temozolomide’s sensitivity and demonstrated that, under standard treatment with temozolomide, EGFRvIII expression leads to prolonged survival only in patients with tumors with O6-methylguanine-DNA methyltransferase (MGMT) methylated promoter. Their results showed that EGFRvIII sensitizes a type of GBM to the current standard of care treatment with temozolomide through the upregulation of DNA mismatch repair (MMR) [65]. However, patients with tumors that have both EGFRvIII and MGMT methylation are very uncommon, and the conclusion that EGFRvIII status was associated with increased survival had a p = 0.06. This level would not normally be considered significant, especially not in this sort of multivariate analysis[66].
The EGFR gene is amplified in approximately 40% of glioblastomas. The primary and secondary GBM differ in genetic profiles and primary GBMs have a higher prevalence of EGFR gene amplification and overexpression than secondary GBMs[67]. In a study performed by Watanabe et al., EGFR gene amplification was associated with protein overexpression in most tumor cells, but 10% of GBM with overexpression of EGFR protein lacked EGFR gene amplification[68]. However, previous studies have stated that EGFR overexpression or activation does not necessarily cause a simple amplification of its downstream signals, but dose-dependent changes in oncogene-induced downstream signaling and biological responses have been reported [69].
Breakpoint sequence analyses proved different types of chromosomal rearrangements and mechanisms of DNA repair. Analyses of single nucleotide polymorphisms suggested that different deletions may appear from amplified non-vIII EGFR precursor[70].
In a study performed in 2018 on glioma tumor samples by Tomoyuki et al., complex chromosomal rearrangements involving chromosome 7 were observed[70].
A study performed by Lopez-Gines et al. showed that trisomy/polysomy 7 and monosomy 10 were frequently associated with glioma. The combination of these anomalies is important in glioblastoma’s tumorigenesis. Moreover, the association seems to be independent of EGFR gene amplification[71].
It is well known that wild-type EGFR ligands such as transforming growth factor-alpha (TGF-alpha) and heparin-binding EGF (HB-EGF) are often increased in glioblastoma leading to an autocrine loop resulting in the autonomy growth of glioma cells[72]. GBM expresses an EGFR mutant (EGFRvIII) that signals constitutively, does not bind ligand, and is considered to have more tumorigenicity than wild-type EGFR. In a U251-MG glioma cell line, the expression of EGFRvIII may result in specific up-regulation of some genes (TGF-α, EPHA2, HB-EGF, IL8, FOSL1, MAP4K4, DUSP6, and EMP1) influencing signaling pathways involved in oncogenesis. TGF-α and HB-EGF (EGFR ligands) induce the expression of EGFRvIII, suggesting that EGFRvIII has a role in creating an autocrine loop with wild-type EGFR. By inhibiting HB-EGF activity with neutralizing antibodies, EGFRvIII-induced cell proliferation may be reduced, suggesting that EGFRvIII-HB-EGF-wild-type EGFR autocrine loop has a major role in signal transduction in glioblastoma cells[73]. Furthermore, studies have demonstrated that the expression of the EGFR alone has a poor transformation effect on cells. Though, coexpression of TGF-α ligand leads to a significant increase in transformation and therapies based on neutralizing the ligands have demonstrated the decreased growth of cells that harbor such loops[74][75].





