1000/1000
Hot
Most Recent
 +1 point
+1 point
Rheumatoid arthritis (RA) is a systemic inflammatory autoimmune disease identified by continuous joint inflammation promoting cartilage and bone damage, incapacity, and eventually systemic complications.
The progression of the disease promotes loss of function, reduced quality of life and increased morbidity and mortality. In addition, RA can also affect organs outside the joints, mainly the lungs, skin, and cardiovascular system.
The precise etiology of RA is still unknown, but genetic and environmental stimuli clearly participate. Dysregulated adaptive immunity can precede the clinical manifestations for many years, and it is likely that the repeated activation of innate immunity can contribute to a breakdown of tolerance.
Regarding the mechanism of RA, it was shown that post-translational modifications in the mucosa (such as citrullination or carbamylation) generate new epitopes recognized by the adaptive immune system. Modified peptides are then presented by APC (antigen-presenting cells) that in turn activate in lymphoid tissues the adaptive immune response and promote autoantibodies formation. Fibroblast-like synovial cells (FLS), macrophages and APCs can be activated in the synovium to produce several inflammatory factors. Next, the autoimmune response caused by the immune system generates synovial inflammation, thus promoting cytokine secretion and synovial blood vessel leakage. Lastly, the autocrine and paracrine effects of cytokines and the continuous adaptive immune response can make the disease permanent and eventually lead to cartilage and bone destruction [1].
In addition, oral infections with specific microorganisms such as Porphyromonas gingivalis have been associated with the RA development. The pathogenic mechanisms involved in the relationship of periodontal disease and RA comprise the increased production of T helper 17 by the microorganisms, the burden of citrullination, the generation of self-antigens that may cross-react with host protein and convert to autoantigens and the possibility of the bacterial translocation to the joints increasing the inflammatory burden leading to the osteoclastic activation, among others. All these mechanisms can trigger the onset of RA and the perpetuation of the inflammation, worsening the progression of the disease (reviewed in Rooney C.M. et al., 2020) [2].
RA is a dissimilar disease, involving multiple clinical manifestations and pathogenic mechanisms among individuals with the same diagnosis and/or throughout different disease stages. Indeed, although autoantibodies are an important feature of RA, several RA patients are negative for these autoantibodies. These traits further support the complexity of the disease and the involvement of numerous factors in the trigger and the evolution of RA (Figure 1).
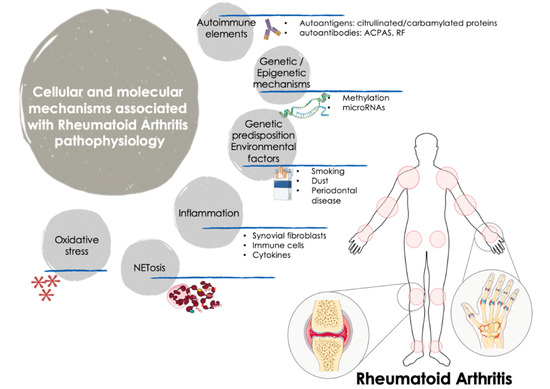
Figure 1. Cellular and molecular mechanisms involved in the pathogenesis of RA. The onset and progression of RA is orchestrated by several cellular and molecular pathologic mechanisms that participate in an integrated and coordinated way. The interaction between multiple genetic and environmental factors increase the risk of RA development. Autoimmune factors such as autoantigens and autoantibodies along with inflammatory mediators released by activated immune cells -including several cytokines and chemokines- are well recognized molecular features that influence the clinical manifestations of RA, characterized by tender and swollen joints and other related comorbidities. In recent years, novel mechanisms have been further associated with the pathogenesis of RA, including oxidative stress and NETosis and the interplay among gene expression and epigenetic and post-transcriptional mechanisms such as methylation and microRNAs. ACPAS, Anti-citrullinated peptide antibodies; RF, Rheumatoid Factor.
Thus, it was widely demonstrated that along with synovium and immune cells, cytokines play key roles in the pathophysiology of the disease, through their involvement in cell growth, proliferation, differentiation, tissue repair and regulation of the immune response, being responsible for inflammation and joint destruction, and even for the systemic involvement in this autoimmune disorder [3].
RA is also related to oxidative stress. In RA patients, reactive oxygen species (ROS) produced by joints and circulating neutrophils, monocytes and macrophages can damage various cell constituents, including carbohydrates, proteins, lipids, and DNA. Accordingly, ROS may induce death of chondrocytes and promote a higher inflammatory damage both at articular and systemic levels. Moreover, a robust positive correlation between free radicals (generated both in synovial fluid and peripheral blood) and DAS28 score was proven, underscoring that ROS can be considered a predictive marker for the inflammatory status in patients with RA [4].
Accelerated NETosis (the process wherein neutrophils release highly decondensed chromatin and granular contents to the extracellular space, called neutrophil extracellular traps -NETs-) was also related to the pathogenesis of RA patients, through the externalization of immunostimulatory molecules and citrullinated autoantigens that produce abnormal adaptive and innate immune responses both in the joint and in the periphery, thus perpetuating damaging mechanisms and fostering the development of related comorbidities in this disease [5].
From the genetic standpoint, recent studies (including both gene-arrays and new generation sequencing studies) uncovered the gene expression signatures and networks in synovial tissue and purified immune cells associated with the pathogenesis of RA [6]. Thereafter, epigenetic mechanisms underlying gene regulation, and documented to be altered in RA were also evaluated. Thus, genome-wide methylation studies identified several changes in FLS and immune cells linked to the origin and the development of this disorder [7]. In addition, the study of post-transcriptional mechanisms of gene regulation identified several microRNAs directly associated with the disease activity, the production of inflammatory mediators and the osteoblastogenesis [8].
The unmet need for new treatment alternatives for RA patients encouraged research in the development of novel drugs regulating physiologically relevant targets. In this context, biologic immunosuppressive therapies (named biologic disease-modifying antirheumatic drugs, bDMARDs) have proven to control disease activity and halt the progressive joint destruction. These drugs were designed to target the inflammatory molecules, cells, and pathways that confer tissue damage in RA patients.
Several autoimmune and cellular constituents can be pondered as key factors for the occurrence and development of RA, including autoantibodies, synovial fibroblasts, and white blood cells such as monocytes, lymphocytes, and neutrophils. The activation of these cells promotes the production of inflammatory mediators and cytokines, thereby perpetuating this pathogenic process.
Most patients with RA will develop anti-citrullinated protein antibodies (ACPAs). Therefore, in the early stages of RA development, tolerance to citrullinated antigens is disturbed, and the anti-citrullinated humoral response was expanded, fostering the development of inflammation and eventually arthritis.
The biological activities of ACPAs include stimulating the production of pro-inflammatory cytokines, inducing osteoclast formation, and promoting the release of autoantigens by neutrophils [9]. As such, in a previous study, we demonstrated that ACPAs purified from RA patients fostered the induction of pro-oxidation, pro-inflammatory and atherosclerotic states of different white blood cell subpopulations [10]. In addition, an effect on macrophage activation and survival was also confirmed. Therefore, ACPA-mediated processes might contribute to the progress and/or prolongation of RA.
FLS are key components of RA infiltrating synovium and play a major role in the occurrence and persistence of destructive joint inflammation. The pathogenic capacity of FLS in RA derives from its capability to secrete immunomodulatory cytokines and inflammatory mediators, as well as multiple matrix modeling enzymes and adhesion molecules [11].
Likewise, monocytes/macrophages play a major role in inflammation by releasing cytokines and infiltrating extensively at sites of inflammation, such as synovium. In addition, given that monocytes/macrophages are ‘over-differentiated’ into osteoclasts, they are linked to bone erosion in RA. Besides, the increase in the number of macrophages in the lower layer of the synovium is an early feature of active rheumatism, so that the presence of high number of macrophages is the main feature of inflammatory lesions. Moreover, the degree of infiltration of synovial macrophage is associated with the degree of joint erosion, and the removal of these macrophages from inflamed tissues has great therapeutic benefits [12][13][14].
Lymphocytes are the most studied cells that mediate the pathogenesis of RA, so that RA has been largely considered to be a disease driven by T helper (Th) 1 cells. Th1/Th2 is imbalanced, being higher the levels of Th1 cells. Th1 secretes inflammatory molecules, including interleukin (IL)-2, interferon-γ (IFNγ), and tumor necrosis factor-α (TNFα), thus preventing the differentiation of CD4+ T cells into Th2 cells [15]. Th17 lymphocytes were also associated with the pathogenesis of RA, mainly producing IL-17A, IL-6 and TNFα. These cytokines are expanded in RA patients’ serum and promote the activation of other cells.
Regulatory T cells (Treg) are also involved in the RA pathology. They play an important role in immunosuppression through cell-cell interactions, and are significantly reduced in the late stage, and show impaired function in RA. Therefore, it was proposed that the development and progression of RA are caused by the imbalance of Th1/Th2 and Th17/Treg cells [15][16].
Equally, B lymphocytes play a fundamental role in the pathogenesis of RA. They are a source of ACPAs and RF, and they contribute to the formation of immune complexes and the stimulation of complement in joints. B cells are also very effective antigen-presenting cells and stimulate the activation of T cells by co-stimulatory molecules. In addition, B cells both produce and respond to chemokines and cytokines, thereby promoting the infiltration of leukocytes into joints, the ectopic lymphoid structures formation, angiogenesis and synovial hyperplasia [3].
Finally, among cells of the immune compartment, neutrophils are the most abundant cells in the synovial fluid of the damaged joints and in the pannus/cartilage interface where tissue injury occurs. The pathogenic role of neutrophils in RA involve changes in several processes, including augmented cell survival and migration, abnormal inflammatory activity, increased oxidative stress and release of NETs. By activating these mechanisms, neutrophils can stimulate other immune cells, thereby perpetuating inflammation and promoting the destruction of injured articular cartilage and bones [17].
Cytokines, together with synovial cells and immune cells, and as products and controllers of the immune system in RA, play vital biological processes, such as cell growth, differentiation, proliferation, inflammation, tissue repair and immune response regulation. This causes inflammation and joint destruction that occurs during arthritis development [18].
Hence, the characteristic inflammation of RA is due to the predominance of pro-inflammatory cytokines over those of anti-inflammatory cytokines.
A myriad of studies supports that the most relevant pro-inflammatory cytokines in the pathogenesis of RA are TNFα, IL-1, -6, -7, -12, -17, -18, -23, and -32. They are mostly produced by myeloid cells, fibroblasts, endothelial cells, keratinocytes and chondrocytes, which are also involved in the production of metalloproteinases, oxidative stress mediators and prostaglandins that lead to inflammation, osteoclast activation and differentiation, osteoclastogenesis, neovascularization and angiogenesis [19].
Instead, several anti-inflammatory cytokines counterbalance the chronic activation of RA immune cells. Thus, Th2 cells and a set of innate lymphoid cells (ILC2s) secrete IL-4 and IL-13. These cytokines induce in macrophages the differentiation into a regulatory M2 phenotype via signal transducer and activator of transcription 6 (STAT6) activation. Then, M2 macrophages release transforming growth factor beta (TGFβ) and anti-inflammatory cytokines such as IL-10. Moreover, IL-5 produced by ILC2s and Th2 cells recruit eosinophils, which shift the balance of macrophages to primarily being regulatory M2 macrophages, thus diminishing the secretion of pro-inflammatory cytokines such as IL1β and TNFα by M1 macrophages [20].
In this complex landscape, it is further proven that leukocyte recruitment is a key process in regulating the onset of inflammation [21]. Throughout the course of life, a subset of white blood cells continues to enter, pass through, and leave the matrix of the bone marrow, thymus, lymph nodes and surrounding tissues.
Patients with RA have defects in several checkpoints regulating leukocyte entry and exit from lymphoid tissues and the joints.
Previous studies have shown that synovial endothelial cells (first barrier that leukocytes must traverse to enter the joint) from RA patients induce the recruitment of leukocytes after TNFα stimulation [22]. Actually, the rheumatoid joint microenvironment was demonstrated to promote the phenotypical alteration of synovial endothelial cells [23]; such cells have increased expression of the adhesion molecules vascular adhesion molecule 1 (VCAM1) and E-selectin together with enlarged expression of both peripheral node addressin (PNAd) and vascular adhesion protein 1 (VAP1) [24]. Thus, leukocyte entry into the join depends on the phenotype of its gatekeepers, the synovial endothelial cells.
Another factor to consider in this tightly regulated process is the self-regulation of leukocyte trafficking. Thus, it was shown that a B cell derived peptide termed PEPITEM, a key constituent of the immunoprotective pathway that reduces the T cell trafficking into inflamed tissues, is absent in patients with RA [25]. Hence, in RA patients, circulating B cells express reduced levels of receptors for adiponectin, do not secrete PEPITEM in response to adiponectin and are thus unable to suppress T cell migration in vitro.
That overall data point at a significant heterogeneity in leukocyte trafficking patterns in RA patients, which are further involved in both the evolvement of the disease and the response to several therapies [26].
The latest type of treatments used for RA therapy is biological compounds or biological response modifiers, designed to target inflammatory molecules, cells, and pathways that cause tissue damage in RA patients.
The bDMARDs currently approved and clinically demonstrated as most effective include TNFα inhibitors (infliximab, etanercept, adalimumab, etc.), T cell costimulation modulators (abatacept), IL-6-inhibitors (tocilizumab, sarilumab), IL-1 inhibitors (anakinra), and anti-CD20 drugs (rituximab). Most of them are direct inhibitors of pro-inflammatory cytokines, while others develop their function upstream of the inflammatory cascade, thereby interfering the activation of T cells (i.e., abatacept) or the depletion of B cells (i.e., ituximab) [27] (Figure 2).
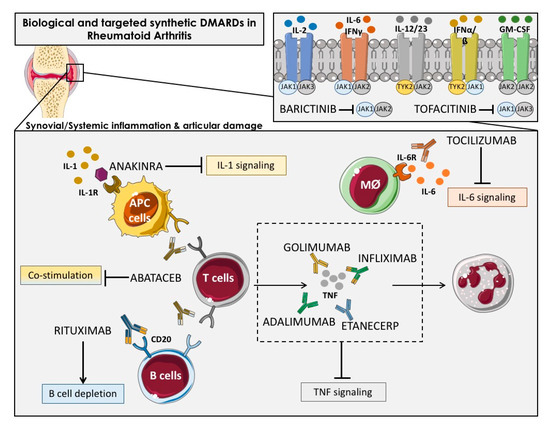
Figure 2. Overview of the main immunologic pathways targeted by the biological and targeted synthetic therapies in rheumatoid arthritis. Simplified description of cytokines and receptors that are targeted by available biologic DMARDs such as: TNF inhibitors (adalimumab, golimumab, infliximab and etanercept), IL-1R antagonist (anakinra), antibody against IL-6R (tocilizumab) and anti-CD20 (rituximab), and targeted synthetic DMARDs, such as Janus kinase inhibitors (anti-JAKs): tofacitinib (JAK1-JAK3 inhibitor) and baricitinib (JAK1–JAK2 inhibitor). APC: Antigen-presenting cells; Mφ: Macrophage.
New encouraging treatments for RA patients encompassed the inhibition of various cytokines by the inhibitors of the Janus kinase family (JAK) [28] including tofacitinib, the first JAK-inhibitor agreed for RA treatment, which inhibits JAK-3 and -1, and baricitinib, which is selective for JAK-1 and -2 [29]. JAK inhibitors, particularly JAK1 inhibitors, can prevent activation of transcription factors of Signal Transducer and Activator of Transcription STAT transcription factors in the synovium and are effective RA treatment drugs. Synovial biopsy studies have shown that the JAK-inhibitor tofacitinib is associated with clinical improvement of RA [30].
Numerous studies, including ours, demonstrated the anti-inflammatory effects of bDMARDs, both in vivo and in vitro, in RA patients [10][31][32].
Thus, several studies showed the capacity of biological therapies to reduce the protein levels of different cytokines and adhesion molecules. Bulur A. et al. reported that after 6 months of treatment with anti-TNF therapy including infliximab, adalimumab and etanercept, the serum levels of IL10, IL-12p40, IFN- and macrophage chemoattractant protein-1 (MCP-1) were reduced [33]. In line with this, Charles P. et al. demonstrated that the treatment with infliximab decreased the serum levels of IL-6, IL-1, GM-CSF, IL-8, p55 sTNFR in 73 RA patients after four weeks of treatment [34]. The reduction of IL-6 after anti-TNF therapy was validated by other studies since its activity is directly associated with TNF-alpha [35][36]. In addition to the decrease of IL-6, our group further demonstrated a reduction of IL17 and TNF-a in 96 RA patients treated with anti-TNF therapy for six months [36]. Other cytokines were diminished after TNF inhibitors in the synovium, including IL-8, MCP-1 [37], and IL1 [38]. We also showed that other biological therapies such as Tocilizumab was able to reduce the plasmatic levels of migration/adhesion molecules such as soluble VCAM and E-Selectin [39]. VEGF-A is another key inflammatory and vascular mediator that was shown to be reduced after tocilizumab treatment in RA patients [40].
Yet, recent reports demonstrated that bDMARD not only exert anti-inflammatory effects, but are involved in several mechanisms underlying the pathogenesis of RA. Thus, it was shown that anti-cytokine therapies systematically target many of the molecules involved in the leukocyte recruitment cascade; for example, TNFα and IL-1β are involved in endothelial cell activation and IL-6 is involved in fibroblast–endothelial cell communication. Adalimumab and adalimumab reduced granulocytes influx into the joints of RA-established patients. Treatment with IFX for two weeks also reduced articular T cell, B cell and macrophage counts in comparison to cell counts in pretreatment synovial tissue samples obtained by biopsy [37][41].
By contrast, treatment with adalimumab did not alter monocyte entry into the joints of patients with RA. Instead, adalimumab was suggested to reduce macrophage and monocyte numbers within the joint by influencing their egress [42]. Unlike etanercept both infliximab and adalimumab reportedly restored the function of peripheral blood Treg cells and increased the number of these cells in the circulation [43][44].
Other DMARDs that affect cytokine production, such as JAK inhibitors can also influence the migratory potential of leukocytes. Peripheral blood neutrophils from patients with RA migrate along an IL-8 chemotactic gradient in a JAK2-dependent manner; in vitro, this process was sensitive to the JAK1–JAK2 inhibitor baricitinib but not the JAK3–JAK1 inhibitor tofacitinib [45].
Up to date, there has been less success targeting many other signaling pathways, including p38 mitogen-activated protein kinase (MAPK), MAPK/ERK kinase, spleen tyrosine kinase and Bruton’s tyrosine kinase [46][47].
Several new therapeutic targets, including bDMARDs directed at the innate immunity, and the prevention of joint destruction and migration (inhibitors of IL-6, granulocyte macrophage colony-stimulating factor (GM-CSF), matrix metalloproteinases, chemokines) are now under study in different phases of clinical trials [48].
Overall, these data suggest that the multiple pathways involved in the clinical features of RA converge in different pathophysiological processes that may be simultaneously targeted by bDMARDs, thus offering many opportunities for therapeutic intervention.





