1000/1000
Hot
Most Recent
 +1 point
+1 point
Among the different mechanisms involved in oxidative stress, protein carbonylation and lipid peroxidation are both important modifications associated with the pathogenesis of several diseases, including cancer. Hematopoietic cells are particularly vulnerable to oxidative damage, as the excessive production of reactive oxygen species and associated lipid peroxidation suppress self-renewal and induce DNA damage and genomic instability, which can trigger malignancy. A richer understanding of the clinical effects of oxidative stress might improve the prognosis of these diseases and inform therapeutic strategies. The most common protein carbonylation and lipid peroxidation compounds, including hydroxynonenal, malondialdehyde, and advanced oxidation protein products, have been investigated for their potential effect on hematopoietic cells in several studies.
Oxidative stress can be defined as an imbalance between the production of reactive oxygen species (ROS) and the ability of the cells to detoxify them [1]. ROS production is a common consequence of aerobic metabolism, and can play a dual role in cells, being either beneficial or harmful. For instance, hydrogen peroxide is a major redox metabolite that operates in redox signaling, but when produced at high concentrations it can contribute to damage of biomolecules and trigger an inflammatory response [2]. High levels of ROS are associated with DNA fragmentation, lipid peroxidation, and/or protein carbonylation, leading to cellular dysfunction and even cell death [3]. Accordingly, cells rely on efficient antioxidant defenses provided by enzymes and metabolites to maintain low levels of ROS; for example, superoxide dismutase (SOD) and catalase (CAT) enzymes, and antioxidant molecules such as thiol antioxidants or vitamin E [3].
As oxidative stress has myriad consequences for cell fate, many analytical procedures have been established both for the produced ROS and their downstream effects on biomolecules. Although free radicals can be measured in biological samples, their quantification lacks sensitivity and specificity, and so much effort has been directed at quantifying their target products, including fragmented DNA, lipid peroxidation products (malondialdehyde (MDA) or 4-hydroxy-2,3-nonenal (HNE)) and protein carbonylation, an irreversible oxidative modification [4]. As DNA fragmentation does not directly correlate with ROS levels, the most useful current methods involve the quantification of lipid peroxidation and protein carbonylation [5].
Protein carbonylation is one of the most common oxidative modifications. Oxidation of proteins is of particular concern since it leads to aggregation, polymerization, unfolding, or conformational changes that may confer a loss of structural or functional activity. Oxidized protein aggregates are not readily degraded in the cell, and their accumulation causes cell dysfunction [6][7]. While many different types of protein oxidative modifications are possible, most involve protein carbonyls (aldehydes and ketones) [8]. As carbonyl groups are chemically stable, they are extremely useful for laboratory analysis, although their study is methodologically complex. The carbonyl contents of individual proteins may be assessed through derivatization of the carbonyl group with dinitrophenylhydrazine (DNPH), which forms a stable dinitrophenylhydrazone (DNP) product that can be analyzed spectrometrically or by immunoblotting [9][10]. Carbonylation research is characterized by the application of numerous protocols and proteomics workflows, which allows the measurement of compounds by several techniques. Protein carbonylation is a major final by-product of multiple oxidation pathways that occur in the cell and thus, this makes it an appropriate marker of oxidative stress [11]. In contrast, some protein modifications might just represent cellular antioxidation mechanisms as part of the oxidation-defense system [12]. To deepen into the biological implication of protein carbonylation, it is important to characterize each specific modification.
Protein carbonylation can be induced directly by the action of oxidative stress or indirectly by reactions of secondary by-products.
The main mechanism of protein carbonylation involves the direct action of ROS or the metal-catalyzed oxidation of amino acid side chains, particularly proline, arginine, lysine, and threonine. Carbonyl derivatives can also be generated through the α-amidation pathway or through the oxidation of glutamyl side chains, where the peptide is blocked in N-terminal amino acids by an α-ketoacyl derivative [13]. The indirect mechanism of protein carbonylation involves the carbonylation of lysine, cysteine, and histidine, which may be caused by their reaction with reactive carbonyl groups produced during the oxidation of carbohydrates (e.g., glyoxal (GO), methylglyoxal (MGO)) and lipids (e.g., HNE, MDA or acrolein (ACR)). This process of carbonyl generation is termed glycoxidation (the formation of advanced glycation end-products (AGEs)) and lipoxidation (the formation of ALEs), respectively [13][14][15][16][17][18]. Advanced oxidation protein products (AOPPs) are modified structures, similar to AGEs, which also serve as oxidative stress markers [19] (Figure 1).
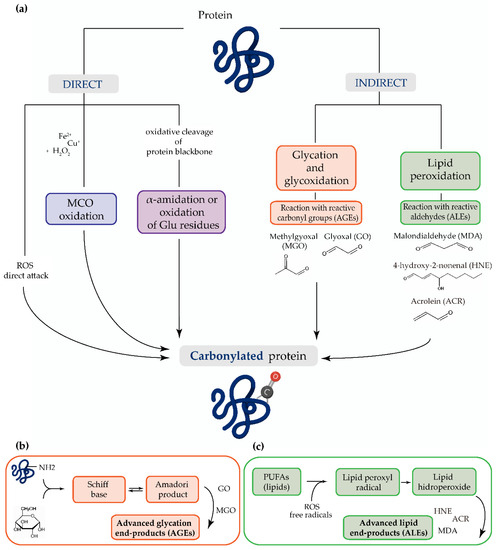
Figure 1. (a) The most common mechanisms of protein carbonylation. Direct processes include reactive oxygen species (ROS) attack, metal-catalyzed oxidation (MCO), and by oxidative cleavage of protein backbone (via the α-amidation pathway or through oxidation of glutamine side chains). The indirect mechanisms involve the reaction with (b) advanced glycation end-products (AGEs) and (c) advanced lipid peroxidation end-products (ALEs).
Lipid peroxidation is also a widely used biomarker of oxidative stress. The polyunsaturated fatty acyls (PUFAs) chains found in membranes and lipoproteins are particularly susceptible to free radical chain autoxidation, leading to a variety of unsaturated lipid hydroperoxides [20]. PUFAs may also be enzymatically oxidized, although these are regio- and stereo-controlled processes involved in normal intermediary metabolism [20]. The nonenzymatic lipoxidation-derived hydroperoxides can decompose, usually in the presence of reduced metals or ascorbate [21], to generate mono- and bifunctional reactive carbonyl-containing moieties, producing aldehydes such as MDA, GO, ACR, 4-HNE, and 4-oxo-2-nonenal (ONE) [20]. The reaction of MDA with thiobarbituric acid to form thiobarbituric reactive substances (TBARS) is a common estimator of oxidative damage. The TBARS assay is, however, nonspecific for MDA, and fatty peroxide-derived decomposition products other than MDA are thiobarbituric acid-positive [20].
The potential effects of ROS as well as their target products on hematopoietic cells are particularly relevant, as these cells are acutely sensitive to oxidative damage associated with the accumulation of free radicals [20]. The resultant lipid peroxidation produced by the excessive production of ROS and reactive nitrogen species can suppress self-renewal, limiting the number of hematopoietic stem cells, and directly induce DNA damage and genomic instability [22].
Lymphoma are a heterogenous group of hematological malignancies derived from different types of lymphocytes and occur predominantly in lymph nodes or other lymphoid structures [23]; as such, they are considered as the solid tumors of the immune system [24]. While their etiology is not well understood, it is likely multifactorial as abnormal genetic alterations, disordered epigenetic regulation, aberrant pathway activation, or infections like Epstein–Barr virus have been reported [23][25][26].
There is growing evidence that oxidative stress and an imbalance in reduction–oxidation might play a significant role in lymphoma carcinogenesis and patient prognosis, either by generating a more favorable environment for cancer cells to proliferate or by modifying the efficiency of oncological treatments, which are largely based on the generation of ROS [27]. Hypoxia is a characteristic feature of solid tumors and, accordingly, its role in hematological malignancies was initially presumed to be inconsequential [28][29]. As mentioned above, however, lymphoma present with solid tumor-like features [30] and normal lymph nodes exhibit low oxygen tension [27][31]. Thus, ROS production might be induced in lymphoma by hypoxic stress, whereas in other hematological diseases it might be due to impaired antioxidant defenses [32].
Multiple myeloma (MM) is the second most common hematological cancer after lymphoma and is characterized by the accumulation of clonal malignant plasma cells in the bone marrow (BM). In fact, the BM microenvironment (niche) plays a key role in supporting tumor cell growth, disease progression, and drug resistance of myeloma plasma cells [33]. One of the main causes of MM is indeed oxidative stress, and it has been known for almost three decades that oxidant/antioxidant parameters are misbalanced in this disease [34] which might continuously stimulate an inflammatory milieu at the tumor microenvironment [35]. The enhanced oxidative state, in turn, increases the rate of genetic mutation, leading to the acquisition of a malignant phenotype and subsequent cancer progression, as also observed in HL [36].
Commonly, MM is preceded by asymptomatic premalignant stages including monoclonal gammopathy of uncertain significance (MGUS) and/or a symptomatic stage such as smoldering multiple myeloma (SMM). Important proteins for the progression of MM, such as c-MYC, have been shown to regulate ROS levels through the modulation of mitochondrial activity [37]. Myeloma cells increase their metabolic demand when the disease progresses, and, consequently, there is a disproportionate production of free radicals or ROS.
Leukemia is known to be a heterogeneous disease and four major subtypes are recognized: acute lymphoblastic leukemia (ALL), chronic lymphoblastic leukemia (CLL), acute myeloid leukemia (AML), and chronic myeloid leukemia (CML). ALL is the most frequent cause of death from cancer before the age of 20 [38] and presents genomic alterations implicated in the proliferation and maturation of lymphoid progenitor cells [39]. CLL is the most common leukemia worldwide and is characterized by the accumulation of B (B-CLL) or T (T-CLL) cells arrested in the early phase of cell division [40], although its ontogeny is unknown [41]. AML is the most common acute leukemia in adults and is defined by the clonal expansion of abnormally differentiated blasts of the myeloid lineage [42]. Finally, CML is a myeloproliferative disorder with a unique genetic rearrangement, the Philadelphia chromosome (BCR-ABL1) which causes the disease [43].
Oxidative stress is a prominent feature in many leukemias [4], and they are characterized by a higher level of ROS than nonleukemic cells. The implication of ROS in the course of leukemias has been scarcely studied [44][32][45][46].
Myelodysplastic syndromes (MDS) are also a heterogeneous group of onco-hematological cell disorders that are characterized by the presence of immature myeloid precursors (blasts), dysplastic hematopoiesis in the BM, and peripheral cytopenias [47][48][49]. Approximately one-third of MDS cases progress to AML [50][51].
Although its origin is not well understood, the role of oxidative stress in the pathogenesis of the MDS have been investigated in several studies [47][52][53][54]. Approximately 60–80% of patients experience symptomatic anemia and 80–90% require red blood cell transfusion support [55]. For this reason, many patients with MDS develop transfusion-dependent iron overload (IOL) [56]. When present in excess, cellular iron leads to toxicity and cell death via free radical formation and lipid peroxidation [57]. It has been reported that the development of IOL significantly worsens the survival of patients with MDS, and it is associated with a higher risk of leukemic transformation [58]. While the role of ROS in MDS is established, the role played by lipoxidation products in the disease and its progression is unclear [9]. High concentrations of the HNE adduct have significant cytotoxic effects on DNA synthesis and mitochondrial activity in leukemic cells, but not in normal hematopoietic precursor cells [59]. However, a recent study examining protein carbonylation in MDS found no significant differences in HNE adducts in BM samples between the MDS and the control group [60]. Analysis of the lipid peroxidation products MDA and nitrite revealed significantly higher levels in patients with MDS and IOL compared with peers without IOL and the control group, and both parameters positively correlated with the levels of ferritin [61] (Figure 2). The same authors later confirmed the increase in MDA levels and higher levels of antioxidant enzymes [22], suggesting that increases in lipid peroxidation is followed by an increase in antioxidant capacity.
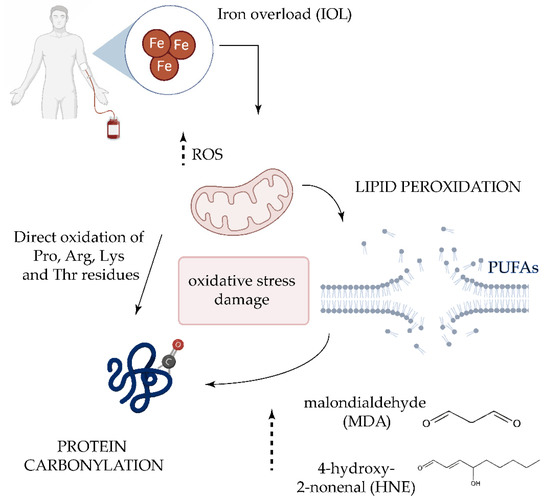
Figure 2. Oxidative damage in transfusion-dependent patients with MDS. Frequent transfusions in patients with MDS leads to iron overload in serum, which plays a key role in the generation of highly reactive oxygen species (ROS) [62]. The increase of ROS could directly oxidize proline (Pro), Arginine (Arg), Lysine (Lys), and Threonine (Thr) residues. ROS-induced lipid peroxidation of long-chain polyunsaturated fatty acids (PUFAs) also promotes protein carbonylation by reaction with lipid peroxidation end-products such as malondialdehyde (MDA) and 4-hydroxy-2-nonenal (HNE).
BCR/ABL negative myeloproliferative neoplasms (MPNs) are unique hematopoietic stem-cell disorders that share mutations that constitutively activate the physiologic signal-transduction pathways responsible for hematopoiesis [63]. MPNs are clonal disorders that are mainly characterized by hyperproliferative BM with varying degrees of reticulin/collagen fibrosis, extramedullary hematopoiesis, abnormal peripheral blood count, and constitutional symptoms. They include polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF) [64].
An imbalanced oxidative status, higher ROS levels, and lower total antioxidant capacity levels compared with controls was found in patients with myelofibrosis by Verner et al. Additionally, the oxidative stress and MDA levels were increased, whereas the total antioxidant status was lower [65]. Following therapy, oxidative stress index and MDA values were significantly lower than the pretreatment values [66]. Higher plasma levels of MDA together with significantly higher protein carbonyls content was also recently reported in patients with MPNs compared with healthy subjects [67].
In patients with PV and ET, Musolino et al. evaluated oxidative stress, finding higher levels of advanced oxidated protein products and S-nitrosylated proteins in both diseases and an increase of AGEs in patients with ET with respect to controls. The authors found a correlation between S-nitrosylated proteins and hemoglobin values in patients with PV, and between AGEs and thrombotic events in patients with ET, suggesting a potential role of ROS in the onset of myeloproliferative-associated thrombotic risk [68].
As discussed earlier, the misbalance of ROS production in cancer cells can alter cell survival mechanisms. Accordingly, a number of studies have focused on developing new treatment strategies by targeting the redox system in tumor cells. Interestingly, while some of the tested agents exert antioxidant properties, a large number of them are also documented to increase the levels of intracellular ROS in addition to blocking a biochemical target [69][70]. Given the large number of targeted therapies tested, we focused only on those agents that have a modulatory effect on oxidative stress, by affecting either protein carbonylation or lipid peroxidation (Figure 3).
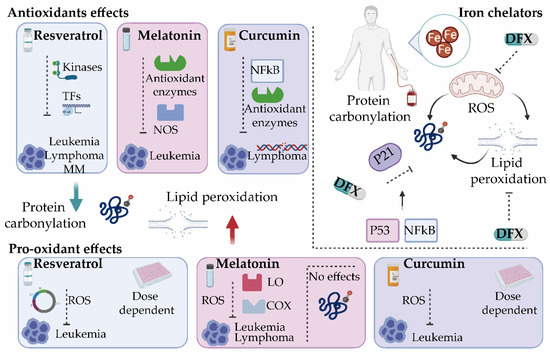
Figure 3. Oxidative stress modulators for the treatment of hematological malignancies. Several antioxidants such as resveratrol, melatonin, and curcumin exert both antioxidant and pro-oxidant activities. By modulating different antioxidant enzymes and transcription factors (TFs), these compounds reduce protein carbonylation and/or lipid peroxidation and inhibit tumor progression. They present cytotoxic effects by enhancing reactive oxygen species (ROS) production. Iron chelators such as deferasirox (DFX) inhibit ROS production directly or indirectly by suppressing the active redox forms of iron and regulating mitochondrial activity and can significantly decrease lipid peroxidation and protein carbonylation in a mechanism dependent on the cell cycle and p21. It will be interesting to explore whether DFX exerts its control on p21 through NF-κB [71] or by inhibiting signaling pathways activated by oxidative stress that control the cell cycle via p53 [72]. COX, cyclooxygenase; LO, lipoxygenase; MM, multiple myeloma; NOS, nitric oxide synthase.
As the total cellular antioxidant capacity is generally compromised in hematologic malignancies, administration of antioxidant drugs might represent a successful way to restore the redox balance. Several antioxidants have been already evaluated as potent anticarcinogenic agents in different kinds of tumors, both in monotherapy or in combination with other antioxidants or classical chemotherapeutics and some of them even revealed promising results in clinical trials. Compared to classical treatment, these compounds possess several important advantages. Beside lower costs they do not exert serious side effects on normal tissues and can be used for chemoprevention [70].
Antioxidants, such as spirulina [73], Enhydra fluctuans extracts [74], and selenium [75][76] can re-establish hematological parameters while simultaneously reducing protein carbonylation and/or lipid peroxidation levels.
In the context of hematological tumors, the antitumor activity of the natural polyphenol resveratrol has been tested in virtually all types of blood cancer cells, including leukemias, lymphomas, and MM [77][78][79][80]. Resveratrol protects phospholipids from oxidation [70], although it is able to inhibit all stage of carcinogenesis (e.g., initiation, promotion, and progression) [81][82][83] by modulating transcription factors, upstream kinases, and their regulators [84]. As resveratrol has no impact on hematopoiesis [85] its potential utility for ex vivo pharmacological purging of leukemia cells from BM autografts before transplantation has been proposed [86][87]. Resveratrol has also been shown to reverse drug resistance in a broad range of in vitro cell systems by sensitizing tumor cells to drug-mediated effects in combination with other chemotherapeutic agents [82]. Nevertheless, a phase 2 clinical trial of resveratrol with or without bortezomib for patients with relapsed and/or refractory MM highlighted side effects and an unacceptable safety profile in combination with bortezomib in these patients [88].
The pituitary hormone melatonin (N-acetyl-5-methoxytryptamine) is of great interest as an endogenous redox modulator with anticancer activity. Melatonin acts directly as a chelator of ROS [89][90][91][92][93], and indirectly by regulating the expression and activities of antioxidant enzymes and nitric oxide synthase [94][95][96][97]. Melatonin has been shown to have a synergistic cytotoxicity effect in combination with lymphoblastic leukemia drugs such as doxorubicin, decreasing ROS and carbonyl formation. Interestingly, this combination is safe for normal lymphocytes, pointing to melatonin as a promising adjuvant for anticancer therapy by allowing lower doses of the anticancer drugs, minimizing their side-effects [98].
In in vitro models of lymphoma, a reduction in lipid peroxidation and/or protein carbonylation products have also been reported using a variety of antioxidants, including 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO), ascorbic acid (vitamin C), α-tocopherol, β-carotene [99], flavonols such as quercetin and rutin [100], canthaxanthin [101] or mangiferin, a C-glycosyl xanthone [102]. Interestingly, extracts of Cocculus hirsutus [103], β-carotene [104], and α-Tocopherol [105] increased survival time in vivo.
In addition, in a murine lymphoma model, ellagic acid inhibits lipid peroxidation and protein carbonylation, decreases PKCα c-Myc expression, and improves TGF-β1 expression in addition to decreasing cell viability, supporting its anticarcinogenic action [106][107].
Another natural product belonging to the group of polyphenols is curcumin or diferuloylmethane, which inhibits free radicals from mediating peroxidation of membrane lipids or oxidative DNA damage [70]. The chemotherapeutic potential of curcumin has been also tested in mice with lymphoma. Results showed that curcumin administration leads to a decrease in lipid peroxidation and protein carbonylation levels, and an increase in the expression and activity of antioxidant enzymes, which in turn modulate the activation of NF-κB, overall reducing lymphoma growth [108].
In conclusion, antioxidants are promising drugs in the management of hematological malignancies. Nevertheless, further studies are necessary in order to confirm their role as anticancer compounds. It should be noted that there are conflicting opinions on the administration of antioxidants during cancer therapy. It is still largely unaccepted by the clinical community as some oncologists believe that it may reduce the effectiveness of chemotherapies, which are mostly based on increasing oxidative stress [36].
In addition to antioxidants, several pro-oxidant drugs capable of modulating cellular ROS contents are currently being tested for their possible use in hematological malignancies. Some of them exert both antioxidant and pro-oxidant properties. Remarkably, the aforementioned antioxidants, melatonin, curcumin, and resveratrol, have also been described as potent pro-oxidants in cancer treatment [70][109][110].
Resveratrol has also dose-dependent pro-oxidant effects, measured as protein carbonylation, which is followed by apoptosis and cell damage [111]. Likewise, Gautam et al. demonstrated that resveratrol induces apoptotic DNA fragmentation in three leukemia cell lines (32Dp210, L1210, HL-60) but not in normal BM cells [87]. The pro-oxidant activity of resveratrol has also been linked to the induction of cell cycle arrest [112]. Resveratrol also suppresses growth of myeloid cells. Lee et al. demonstrated that resveratrol inhibits proliferation of promyelocytic leukemia cells and nonmalignant B-cell lymphoblastoid cells by blocking cell cycle progression in G0/G1, and also induces apoptosis in promyelocytic leukemia cells and acute lymphocytic leukemia cells [113][114]. Interestingly, leukemic lymphoblasts isolated from pediatric patients with ALL undergo apoptosis when treated with resveratrol [70][115].
Melatonin seems to stimulate the production of ROS in human myeloid HL-60 cells, eliciting cytotoxic effects [116]. It also increases the activity of lipoxygenases and cyclooxygenase and promotes the production of ROS in Burkitt lymphoma BL41 cells [117]. Similarly, it enhances cell death in Jurkat leukemia cells via a pro-oxidant pathway [118]. When used on tumoral leukocytes, melatonin produces a rapid and transient stimulation of intracellular ROS, but does not lead to oxidative stress, as revealed by absence of protein carbonylation and the maintenance of free thiols [119][120].
Curcumin also shows pro-oxidant anticarcinogenic mechanisms that are concentration dependent: whereas low concentrations decrease ROS production in human leukemia cells, higher concentrations have the opposite effect and favor ROS generation, measured as increased MDA levels [121]. Curcumin exerts cytotoxic activity on human T-cell leukemia cells without affecting normal cells [122]. Mechanistically, curcumin seems to affect histone acetyltransferase [123] and thioredoxin reductase, converting the latter into a pro-oxidant [124]. Moreover, curcumin induces an increase in GSH levels, responsible for the induction of an apoptotic death pathway in lymphoid Jurkat cells [70][125].
Despite the promising anticancer effects of these natural products, contradictory results responsible for its dual effects are continuously described [126]. Chemically unstable structure, feeble pharmacokinetic, and low bioavailability are reasons for the broad bioactivity profile of these compounds, blocking them from reaching maturity as a drug lead. Moreover, the potential health benefits are still questioned. It is important to highlight there is an urgent need to better characterize the polypharmacology of their degradation products. The lack of chemical standardization of potential drugs like resveratrol and curcumin limits an adequate control of biological assays, leading to unpredictable or potentially irreproducible results [127][128].
A novel redox active mediator that selectively targets tumor cells is motexafin gadolinium, which is a synthetic compound that directly inhibits the activity of Trx and protects against protein carbonylation and lipid peroxidation by inducing apoptosis in malignant cells through oxidative stress. Motexafin gadolinium is being tested clinically for the treatment of lymphoma (NCT00089284, NCT00086034) and leukemia [129][130][131].
Arsenic trioxide is used successfully for the treatment of APL, and both induction and consolidated therapy have resulted in complete remission. Kumar et al. demonstrated that arsenic trioxide induces significant oxidative stress (lipid peroxidation), DNA damage, and caspase 3 activity in HL-60 cells in a dose-dependent manner, and reduces GSH levels [132]. It also activates the intrinsic pathway of apoptosis by modulating the translocation of apoptotic molecules such as Bax and cytochrome c and decreasing the mitochondrial membrane potential.
Finally, targeting copper in cancer cells can also serve as an effective anticancer strategy. Copper is an important metal ion associated with the chromatin DNA. Unlike normal cells, cancer cells have elevated copper levels, which play an integral role in angiogenesis. The interaction between Cu(II) and the phytoestrogen coumestrol in lymphocytes results in lipid peroxidation, protein carbonylation, DNA fragmentation, and apoptosis [133].
These data reinforce the necessity of studying the role of oxidative stress modulating compounds in hematological tumors, characterizing not only their pro/antioxidant effects, but also their molecular mechanism. Although the anticancer potential of some natural products is controversial, some compounds (e.g., motexafin gadolinium) present promising results. Moreover, the clinical efficacy of these agents would be assessed by using biomarkers such as carbonylation and lipid peroxidation.
Iron homeostasis is an effective target in the treatment of different hematological tumors, particularly MDS. As discussed earlier, most patients with MDS develop transfusion dependence and IOL, which has a negative impact increasing oxidative stress parameters [9][53][134][135][136]. In this setting, iron chelation, mainly by Deferasirox (DFX) appears to improve survival in patients with lower-risk MDS and in stem cell transplant settings [135][137]. Moreover, it has been shown to reduce mortality and cytopenia and improve the hematological response [9][138][139][140][141].
Deferasirox is an iron chelator commonly used as a treatment in patients with MDS relying on blood transfusions [142]. DFX is a powerful NF-κB inhibitor in myelodysplastic cells acting independently of cell iron deprivation by chelation, and ROS scavenging [71] and the inhibition of the de novo generation of free radicals through the suppression of the active redox forms of iron [62]. DFX constrains ROS damage in hematopoietic progenitor cells by activating transcription factors and mitochondrial biogenesis [143], the dysfunction of which has been observed in cases of MDS with IOL [144]. Interestingly, a significant decrease in the mean levels of ROS and membrane lipid peroxidation has been reported during DFX therapy [145][146]. In addition, patients under DFX treatment have lower levels of protein carbonylation in BM with respect to untreated patients, which is accompanied by a reduction in the expression of the p53 target gene, p21 [60]. It would be interesting to explore whether DFX exerts its control on p21 through NF-κB [71] or, alternatively, by inhibiting signaling pathways activated by oxidative stress that control the cell cycle via p53 [60][72].
Despite the paucity of studies investigating the beneficial effects of iron chelation on protein carbonylation in other hematological diseases, its effect on ROS levels, and thus, on disease control seem robust.
Iron chelating therapy in myeloid leukemias induces the differentiation of leukemia blasts and normal BM precursors into monocytes/macrophages, in a manner involving the modulation of ROS expression [147]. Moreover, the cytotoxic effects of iron chelating therapy on myeloid blasts has been reported in vitro, in vivo, and ex vivo [148][149][150][151][152][153], presenting a synergistic effect with AML drugs such as decitabine and 5-azacytidine [154][155]. In contrast to the effects of decitabine, DFX decreases the ROS levels to varying degrees [154]. Human studies have demonstrated a protective role of DFX after allogenic-hematopoietic stem cell transplantation in AML [156][157] and in a patient with chemotherapy-resistant AML [158].
DFX has also been reported to inhibit mantle cell lymphoma cell proliferation [159][160], and iron deprivation is cytotoxic to malignant B- and T-cells [161][162]. Moreover, in ALL and T-cell lymphoma, DFX displayed synergistic activity with three ALL-specific drugs: dexamethasone, doxorubicin, and L-asparaginase. Iron chelation appears to act through a ROS-dependent DNA damage response and potentiates the action of an inhibitor of the PARP pathway of DNA repair [163].
Finally, in the context of MM, chelation of intracellular iron induces cell death in myeloma cells [164]. Deferasirox also induces apoptosis in MM cells by targeting oncogenic Pyk2/β-catenin signaling [165]. Conversely, iron loading impairs cell proliferation in MM and increases the efficacy of bortezomib, as iron causes lipid oxidation and inhibits proteasome function [166].





