1000/1000
Hot
Most Recent
 +1 point
+1 point
The Kelch-like ECH-associated protein (KEAP1)– Nuclear factor erythroid-derived 2-like 2 (encoded by the Nfe2l2 gene; NRF2) system attracts extensive interest from scientists in basic and clinical cancer research fields, as NRF2 exhibits activity as both an oncogene and tumor suppressor, depending on the context. Especially unique and malignant, NRF2-addicted cancers exhibit high levels of NRF2 expression. Somatic mutations identified in the NRF2 or KEAP1 genes of NRF2-addicted cancers cause the stabilization and accumulation of NRF2. NRF2-addicted cancers hijack the intrinsic roles that NRF2 plays in cytoprotection, including antioxidative and anti-electrophilic responses, as well as metabolic reprogramming, and acquire a marked advantage to survive under severe and limited microenvironments. Therefore, NRF2 inhibitors are expected to have therapeutic effects in patients with NRF2-addicted cancers. In contrast, NRF2 activation in host immune cells exerts significant suppression of cancer cell growth, indicating that NRF2 inducers also have the potential to be therapeutics for cancers. Thus, the KEAP1–NRF2 system makes a broad range of contributions to both cancer development and suppression. These observations thus demonstrate that both NRF2 inhibitors and inducers are useful for the treatment of cancers with high NRF2 activity.
Our body is constantly exposed to a variety of chemical stresses from external environments, including natural ultraviolet radiation, air pollutants from urban industries and water pollutants from micro/nanoplastic particles [1]. Our personal environments, including lifestyle and food, drinking water, tobacco, alcohol and drug consumption, increase the risk of taking in toxic chemicals that produce oxidative and electrophilic stresses and damage macromolecules, such as nucleic acids, proteins and lipids [2]. However, our body is equipped with the ability to eliminate these toxic xenobiotics and adapt to the environment, which we refer to as the adaptive response or the environmental stress response [3][4]. Environmental stresses often perturb intracellular conditions that challenge our body’s homeostasis at the levels of genes, proteins and metabolites, but the adaptive response protects our body from these challenges. In fact, the adaptive response is essential for our survival in modern society. To attain the adaptation ability to overcome the toxicity of environmental xenobiotics and recover a steady state, our body needs to sense the stresses and convert them into intra- and intercellular signals.
Nuclear factor erythroid-derived 2-like 2; encoded by the Nfe2l2 gene (NRF2) is a transcription factor that controls the environmental stress response by changing gene expression profiles [5]. NRF2 regulates a subset of target genes that mainly encode cytoprotective enzymes/proteins critical to the antioxidative response and detoxication [6] (Figure 1A). NRF2 is activated when cells are exposed to oxidative stresses or toxic chemicals (many of which are electrophilic) [7][8]. This activation of NRF2 is fine-tuned by Kelch-like ECH-associated protein (KEAP11), an adaptor for Cullin 3 (Cul3)-based ubiquitin E3 ligase [9] (Figure 1B, left). KEAP1 binds NRF2 and promotes the ubiquitination of NRF2. Under steady-state conditions, ubiquitinated NRF2 is rapidly degraded by the 26S proteasome. NRF2 has a very short half-life, which is less than 20 min [9][10]. Therefore, NRF2 does not exist abundantly under basal conditions, and available lines of evidence support the contention that NRF2 exists at a relatively low level in most organs or tissues. Consistent with this assertion, juvenile NRF2-knockout mice and rats do not show apparent external phenotypes, except for white teeth [11][12][13].
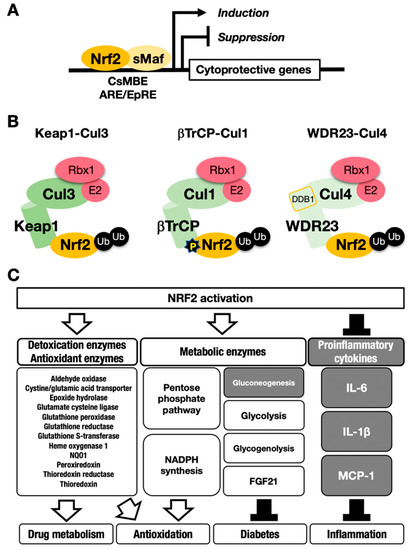
Figure 1. The NRF2-sMaf heterodimer regulates cytoprotective gene expression through the CNC-sMaf-binding element (CsMBE) motif. (A) NRF2 forms a heterodimer with sMaf and binds to the CsMBE motif in the nucleus, which is classically referred to as an antioxidant responsive element (ARE)/electrophile responsive element (EpRE) motif. NRF2 positively regulates genes encoding detoxicating enzymes and antioxidative enzymes and negatively regulates genes coding for proinflammatory factors. Ub, ubiquitin. P, phosphorylation. (B) The combination of an E3 ligase and the adaptor for the degradation of NRF2 through ubiquitination: KEAP1–Cul3, βTrCP–Cul1 and WDR23-Cul4 complexes. (C) NRF2 target genes responsible for the drug metabolism, antioxidation, antidiabetes and anti-inflammation. White arrows indicate induction; black bars indicate suppression.
Ubiquitin–proteasome system-based protein degradation is evident in a number of important regulatory systems [14]. For instance, an inhibitor of the nuclear factor-κB (IκB)-NFκB system, hypoxia-inducible factor (HIF) system and estrogen receptor-α (ERα) system are known to function in response to ubiquitination by specific ubiquitin E3 ligases (e.g., Cul1, Cul2 or Cul4B) and corresponding adaptors. Of the ubiquitin–proteasome-based regulatory systems, the KEAP1–NRF2 system is unique in that it can sense oxidative and electrophilic stresses through the reactive cysteine residues within KEAP1 and mediates the expression of cytoprotective enzyme genes through NRF2 activity. In this system, KEAP1 acts as a sensor for stress, and NRF2 acts as a transcription factor that activates cytoprotective gene expression. Once our bodies are exposed to reactive oxygen species (ROS) or electrophilic toxicants, reactive cysteine residues in KEAP1 are covalently modified by ROS or electrophiles, which stops NRF2 ubiquitination. The activity of modified KEAP1 is weakened, resulting in newly transcribed NRF2 escaping ubiquitination and proteasomal degradation.
This process leads to NRF2 accumulation at the protein level, which induces robust transactivation of cytoprotective genes [7][8] (Figure 1C). NRF2 upregulates the expression of genes encoding detoxicating enzymes and antioxidative enzymes such as NAD(P)H:quinone oxidoreductase 1 (Nqo1) and heme oxygenase-1 (Ho-1, encoded by the Hmox1 gene).
In contrast, NRF2 downregulates genes encoding proinflammatory factors such as interleukin-6 (IL6) and interleukin 1β (IL1β) [15]. NRF2 acts as a transcription factor by forming a heterodimer with a small Maf protein (sMaf), including MafF, MafG or MafK, and by binding to the CNC-sMaf-binding element (CsMBE) (Figure 1A), which is classically defined as an antioxidant responsive element (ARE) or electrophile responsive element (EpRE) [6]. The rapid degradation of NRF2 under normal conditions and quick stabilization upon exposure to stresses allows quick and urgent responses to ROS and/or toxic chemicals (often electrophiles). These are critical features utilized commonly in the environmental stress response systems. Typical examples of such environmental stress response systems are the systems regulated by transcription factors NRF2 and HIF. The activation is evoked by the derepression that halts the repression by constant proteasomal degradation of effector transcription factors.
As NRF2 is regulated at the posttranscriptional level and as NRF2 is an unstable protein, NRF2 activation has been monitored by several indirect approaches, such as monitoring the expression of NRF2 target genes or knocking in fluorescent proteins. Of the NRF2 target genes, Nqo1 encodes a representative marker enzyme that has been exploited for the evaluation of NRF2 activity [11]. In NRF2-induction experiments utilizing cell culture systems, Nqo1 mRNA is usually highly upregulated approximately 12 h and longer after exposure to electrophilic NRF2-inducers [16]. The other representative target genes of NRF2 are glutamate-cysteine ligase catalytic or modifier subunits (Gclc or Gclm) for glutathione (GSH) synthesis [14] and show a similar response profile as Nqo1 mRNA. In contrast, while the Ho-1 gene is also an important NRF2 target, upon induction, Ho-1 gene expression peaks approximately 3 h after a challenge by electrophilic inducers. It has been reported that the Ho-1 gene is under complex regulation, which may elicit the early induction peak [16]. In fact, the upregulation of Ho-1 mRNA levels was not observed in liver-specific KEAP1-knockout mice, even though Nqo1 mRNA levels were upregulated and reflected the NRF2 activation level [17].
NRF2 protein is degraded through the ubiquitin–proteasome system. On the other hand, when we focus on the turnover of KEAP1, it has been shown that KEAP1 is also ubiquitinated in a Cul3-dependent manner, but the 26S proteasome system does not degrade KEAP1 [18]. Furthermore, when cells are exposed to an electrophilic NRF2-inducer, such as tert-butylhydroquinone (tBHQ), the KEAP1 protein is ubiquitinated, and its level is decreased [18]. Therefore, it was concluded that KEAP1 is degraded in a “proteasome-independent” manner. However, it took several years after the paper was published to clarify the molecular mechanism underlying KEAP1 protein turnover.
A clue for this question emerged from the study of p62 (the Sqstm1 gene product). In autophagy-impaired livers, a massive accumulation of p62 is routinely observed concomitant with NRF2 activation. When we examined these livers, we accidentally found abundant KEAP1 protein accumulation [35], suggesting that KEAP1 is degraded through the autophagy pathway. As p62 is a scaffold or chaperone protein in autophagy [36], the most plausible scenario is that p62 binds to KEAP1 and brings KEAP1 to the autophagosome. We have demonstrated this process using several approaches. In the p62-knockout mouse liver, KEAP1 mRNA levels are unchanged, but KEAP1 protein levels are increased, suggesting that KEAP1 is stable without p62 guidance to autophagosomes. p62 binds to KEAP1 through an STGE motif (amino acids 349–352 in mouse/351–354 in human) in the (KEAP1-interacting region (KIR), and phosphorylation of the STGE motif (i.e., pSTGE) increases the binding affinity as the pSTGE motif mimics the ETGE motif in NRF2 [37]. The binding of p62 to KEAP1 through the pSTGE motif results in the inactivation of the ubiquitin ligase activity of KEAP1, indicating that, in impaired autophagy, p62 accumulation activates NRF2 due to the disruption of KEAP1 binding to NRF2. In fact, starvation-induced autophagy accelerated the degradation of the KEAP1 protein [19].
These extensive observations support the notion that KEAP1 is selectively degraded through autophagy, but not the proteasome. The degradation of specific proteins through autophagosomes is referred to as selective autophagy. Thus, the KEAP1–NRF2 system is regulated by two protein degradation systems: the ubiquitin–proteasome system and the selective autophagy system (Figure 2).
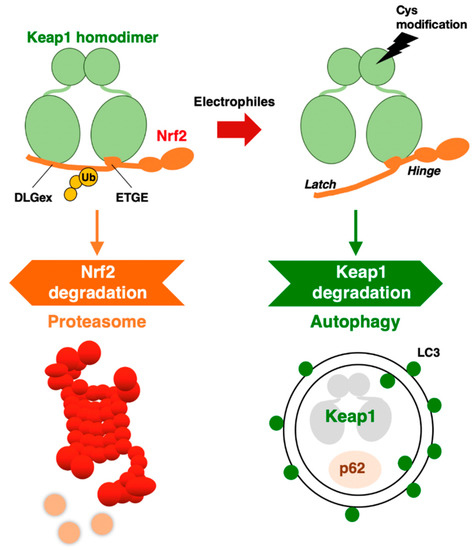
Figure 2. KEAP1–NRF2 system: protein regulation by both proteasome and autophagy.
An intriguing finding along this line is that while the normal half-life of KEAP1 degradation is 12.7 h, the half-life of its degradation is shortened to 3.4 h by tBHQ exposure [19]. In addition to tBHQ, other electrophilic NRF2 inducers that modify KEAP1 cysteine residues, such as diethyl maleate (DEM) and 1,2-naphthoquinone, have also been shown to accelerate KEAP1 degradation. Importantly, the proteasome inhibitor MG132 was reproducibly ineffective at the KEAP1 protein level [18][19], indicating that the 26S proteasome is not critical for KEAP1 degradation.
There are a tremendous number of reports that show somatic mutations in the KEAP1 and NRF2 genes in cancers that originated in various tissues, and their frequencies are especially high in non-small cell lung carcinomas [20][21]. The DLGex and ETGE motifs of NRF2 are two hot spots of somatic mutations in the NRF2 gene, and this observation must have some mechanistic implications [22]. Somatic mutations in the KEAP1 or NRF2 genes cause constitutive NRF2 activation through disruption of the protein–protein interaction (PPI) between KEAP1 and NRF2. In addition to these mutations, oncometabolites [23][24], exon skipping [25], and promoter methylation [26] lead to constitutive NRF2 activation in cancers [14][27]. Furthermore, NRF2 gene expression is transcriptionally regulated by oncogenes, such as K-Ras and c-Myc [28]. Cancer cells with these somatic mutations and the resulting high levels of NRF2 activity are referred to as NRF2-activated or NRF2-addicted cancers, which retain malignant growth with increased proliferation ability and potentiated resistance to chemo- and radiotherapy [29]. To treat these NRF2-addicted cancers, NRF2 inhibitors are needed that exert therapeutic effects [22].
When designing effective treatments for NRF2-addicted cancers, various synergistic and additive interactions of NRF2 with other regulatory factors need to be considered. For instance, PTEN, a tumor suppressor that negatively regulates the PI3K-Akt pathway, upregulates NRF2 activity [30]. Loss of PTEN function in cancers activates Akt phosphorylation of downstream factors, including GSK3β. In 80% of human PTEN-deficient endometrioid tumors, NRF2 is overexpressed in accordance with HO-1 upregulation [30]. Consistently, experiments with cell cultures and mouse models revealed that loss of PTEN activates NRF2 [30][31].
Similarly, NOTCH3 (neurogenic locus notch homolog protein 3) was recently found to play an important role in carcinogenesis under the regulation of NRF2 [32]. NRF2 directly upregulates NOTCH3 mRNA expression, and both NRF2- and NOTCH3-positive cancers show poor prognosis. These observations indicate that there are multiple pathways to consider when developing NRF2 inhibitors. A precise and deep understanding of the molecular mechanisms that regulate NRF2 activation is critical for the development of drugs targeting NRF2-addicted cancers. Some representative small molecule NRF2 inhibitors are shown in Figure 3.
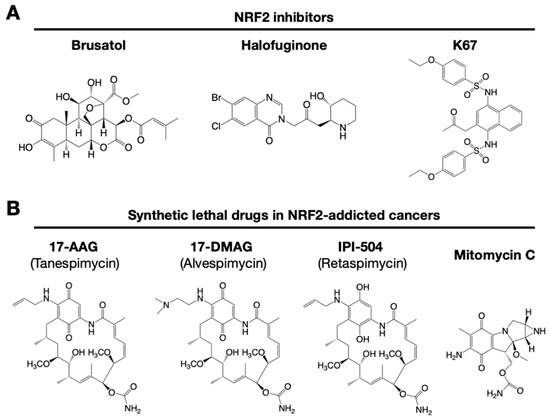
Figure 3. Small chemical inhibitors targeting NRF2. (A) NRF2 inhibitors: brusatol, halofuginone and K67. (B) Synthetic lethal drugs for NRF2-addicted cancers: 17-AAG (tanespimycin), 17-DMAG (alvespimycin), IPI-504 (retaspimycin) and mitomycin C.
It has been reported that brusatol enhances the efficacy of chemotherapy by inhibiting NRF2 [33] (Figure 3A). Brusatol provokes the rapid and transient inhibition of NRF2 through a KEAP1-independent posttranscriptional mechanism [34]. The mechanism appears to be not through direct NRF2 inhibition but through inhibition of protein translation [35]. This action of brusatol overcomes chemoresistance in cancer cells. By inhibiting NRF2, brusatol sensitizes cells to chemical stress provoked by 2,4-dinitrochlorobenzene (DNCB), iodoacetamide (IAA), and N-acetyl-p-benzoquinone imine (NAPQI), the hepatotoxic metabolite of acetaminophen. Whereas brusatol serves as a valuable experimental tool for inhibiting NRF2, the risks presented by its therapeutic use need to be considered, especially its potential for enhancing the sensitivity of nontargeted cells.
Using high-throughput screening of a chemical library, febrifugine was found to inhibit NRF2 activity [36]. Halofuginone is a racemic halogenated febrifugine derivative that was artificially synthesized as a less toxic compound [37]. Halofuginone represses global protein synthesis via the amino acid starvation response elicited by the inhibition of prolyl-tRNA synthetase. As NRF2 is a very short-lived protein even in NRF2-addicted cancer cells, blocking general protein synthesis halts NRF2 accumulation. As an NRF2 inhibitor, halofuginone enhances the chemosensitivity of cancer cells by suppressing NRF2 accumulation.
Accumulated P62 and LC3-II (microtubule-associated protein 1A/1B-light chain 3) are typical markers of impaired autophagy. Phosphorylated P62 accumulates in hepatitis C virus-positive hepatocellular carcinoma (HCC) [38]. In hepatitis and cirrhosis, which are pre-HCC diseases, simultaneous accumulation of P62 and KEAP1 does not frequently occur [39]. However, both P62- and KEAP1-positive lesions are detected in approximately 25% of human HCC and adjacent tissues. The P62 expression level is positively correlated with high levels of NRF2 and NQO1 expression in cultured human HCC lines.
Liver-specific Pten-knockout mice (Ptenflox/flox: Albumin-Cre; Pten-Alb mice) are liver disease models used to develop steatosis, nonalcoholic steatohepatitis (NASH) and liver cancers stepwise [40]. Showing very good agreement with the mouse model, human NASH shows a decreased expression of PTEN mRNA compared to that in normal human liver [41]. In Pten-Alb mice, p62 accumulation elevates the NRF2 level, at least partially, and NRF2 target genes are upregulated [31]. These results imply that, in human cases, accumulated P62 may be a therapeutic target in PTEN-decreased NASH and HCC.
K67 (2-acetonyl-1,4-bis[(4-ethoxybenzenesulfonyl)amino]naphthalene) is an analog of compound 16 (Figure 3A). K67 is an NRF2 inhibitor that disturbs the PPI formed by KEAP1 and P62 phosphorylated at Ser349 in humans (S351 in mice) [42]. K67 effectively inhibits cellular proliferation in HCC, expressing highly phosphorylated P62. Moreover, novel K67 derivatives inhibited the interaction of KEAP1 and phosphorylated P62 [43]. These derivatives increase the sensitivity of cancer cells to anticancer drugs, such as the tyrosine kinase inhibitors sorafenib or regorafenib. These results suggest that K67 derivatives have the potential to be chemosensitizers by inhibiting NRF2 and the expression of NRF2 target genes.
NRF2-addicted cancers show constitutively upregulation of NRF2 target genes. A cell culture system was established in which KEAP1-deleted cells and KEAP1-expressing normal cells were cocultured, and their proliferation was monitored by the distinct colors of fluorescence [44]. Drug screenings that aim to identify synthetic lethal chemical compounds that specifically kill cancer cells with intrinsically high NRF2 activity have been identified. Three geldanamycin-derived heat shock protein 90 (HSP90) inhibitors are synthetically lethal to NRF2-expressing cells (Figure 3B): 17-AAG (17-N-allylamino-17-demethoxygeldanamycin; tanespimycin), 17-DMAG (17-dimethylaminoethylamino-17-demethoxygeldanamycin; alvespimycin), and IPI-504 (the hydroquinone form of 17-AAG, retaspimycin). These two benzoquinone-containing compounds, i.e., 17-AAG and 17-DMAG, are converted into hydroquinones in NRF2-addicted cancer cells in which the expression of drug-metabolizing enzymes is specifically upregulated.
In a similar screening, mitomycin C was found to be a synthetic lethal compound in cells with high NRF2 activity [27]. Quite intriguingly, 17-AAG and mitomycin C exert synergistic effects. Thus, geldanamycin-based compounds and mitomycin C are candidates for drug repositioning to target currently undruggable NRF2-addicted cancers. The use of these drugs in NRF2-addicted cancers may avoid unexpected side effects to normal cells to the greatest extent possible.





