1000/1000
Hot
Most Recent
 +1 point
+1 point
Aminoacyl-tRNA synthetases (ARSs) are essential enzymes that ligate amino acids to tRNAs and translate the genetic code during protein synthesis. Their function in pathogen-derived infectious diseases has been well established, which has led to the development of small molecule therapeutics. The applicability of ARS inhibitors for other human diseases, such as fibrosis, has recently been explored in the clinical setting. There are active studies to find small molecule therapeutics for cancers.
Aminoacyl-tRNA synthetases (ARSs) are a family of 20 essential enzymes that ligate amino acids to their corresponding tRNAs and translate the genetic code during protein synthesis [1]. The two-step catalytic reaction of ARSs involves the formation of an enzyme-bound aminoacyl-adenylate (AMP-aa) followed by the formation of an aminoacylated tRNA (tRNA-aa) by transferring the amino acid to the corresponding tRNA. This catalytic reaction of ARSs plays a pivotal role in protein synthesis, which is essential for the growth and survival of all cells (Figure 1). ARSs have long been studied as therapeutic targets for pathogen-derived infectious diseases, predominantly because of concerns that ARS catalytic site inhibitors would block translation in normal cells [2][3][4]. In this regard, various seminal reviews have discussed ARS inhibitors for the development of antibiotics from different perspectives [5][6][7][8][9].
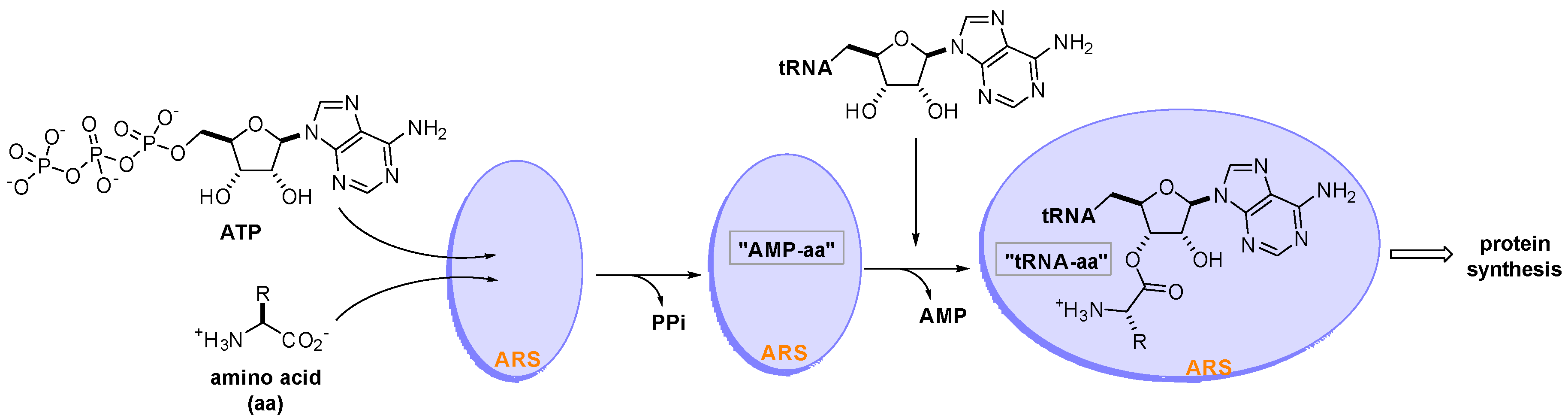
Figure 1. Two-step catalytic reaction of aminoacyl-tRNA synthetases (ARSs). ARSs use an amino acid (aa), ATP, and tRNA as substrates to produce an aminoacylated tRNA (tRNA-aa) that leads to protein synthesis in the ribosome.
However, a significant amount of research targeting mammalian ARSs for the treatment of human diseases have recently emerged. For example, febrifugine derivatives, including halofuginone which targets prolyl-tRNA synthetase (ProRS), are under investigation for the treatment of cancer, fibrosis, and inflammatory diseases [10][11]. Additionally, threonyl-tRNA synthetase (ThrRS) was identified as a target protein of antiangiogenic borrelidin [12], and a peptide fragment derived from tryptophanyl-tRNA synthetase (TrpRS) was studied as a possible anti-angiogenic agent [13]. However, Pfizer has dropped its development of the TrpRS fragment as an anti-angiogenic lead a while ago. Accordingly, Kwon et al. [14] separately provided thorough reviews on the physiology, pathology, and structural aspects of ARSs, as well as the development of ARS inhibitors for human diseases, including infectious diseases, cancer, inflammatory diseases, and central nervous system (CNS) disorders.
Benzoxaboroles are a class of boron-containing compounds with a broad range of biological activities. A subset of benzoxaboroles have antimicrobial activity primarily due to their ability to inhibit leucyl-tRNA synthetase (LeuRS) via the oxaborole tRNA-trapping (OBORT) mechanism, which involves the formation of a stable tRNALeu−benzoxaborole adduct in which the boron atom interacts with the 2′- and 3′-oxygen atoms of the terminal 3′ tRNA adenosi006Ee [15]. In a related study, researchers at Anacor Pharmaceuticals reported an X-ray crystal structure of Thermus thermophilus LeuRS complexed with tRNALeu and the benzoxaborole-based LeuRS inhibitor AN2690 to form a tRNA–AN2690 adduct possessing tetrahedral spiroborate structure 1 (Figure 2) [16]. The tetrahedral spiroborate adduct was stabilized by the formation of covalent bonds between the Lewis acidic boron atom and the 2′- and 3′-oxygen atoms of the tRNA's 3′-terminal adenosine as a nucleophile [17][18]. The oxaborole scaffold can reversibly form covalent tetrahedral complexes with nucleophiles, such as hydroxyl groups, because of the presence of the heterocyclic boron atom, which has an empty p orbital. Targets for these compounds include β-lactamases, cyclic nucleotide phosphodiesterase 4 (PDE4), Rho-associated protein kinase (ROCK), carbonic anhydrase, and LeuRS [19].
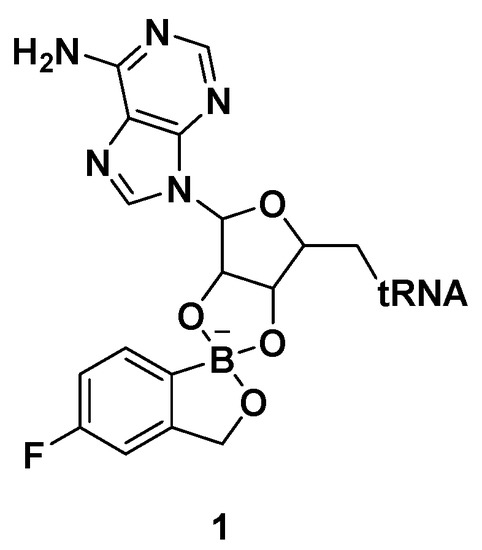
Figure 2. AN2690-tRNALeu adduct via the oxaborole tRNA-trapping (OBORT) mechanism.
A recent example of the benzoxaborole class of inhibitors is the nitrophenyl sulfonamide-based benzoxaborole PT638; Si et al. investigated PT638 to determine its mode of action [15]. The finding that PT638 has potent antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA) (MIC [minimum inhibitory concentration] of 0.4 μg/mL) and does not inhibit Staphylococcus aureus LeuRS (Sa LeuRS) (IC50 [the half maximal inhibitory concentration] >100 μM) suggests the possibility that it has targets other than LeuRS. The selection for resistance to PT638 using MRSA and whole-genome sequencing of three colonies that showed MICs of ≥3.125 μg/mL resulted in three mutations in the LeuRS gene, including D343Y, G302S, and F233I, all of which are located within the editing domain of Sa LeuRS, thereby confirming Sa LeuRS as the target of PT638. A metabolism study of PT638 using MRSA cell lysate and a SAR (structure-activity relationship) study of PT638 analogs revealed that PT638 might be a prodrug that can be reduced to its amine analog PT662 mainly by nitroreductase NfsB in MRSA (Figure 3).
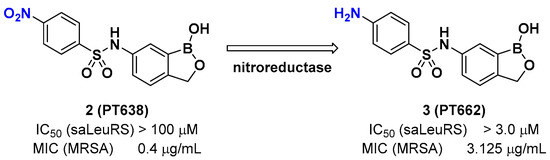
Figure 3. Benzoxaborole PT638 is reduced by a nitroreductase to PT662.
In a related study, GlaxoSmithKline (GSK) developed a series of 3-aminomethylbenzoxaborole derivatives 4 and 5 that target Mycobacterium tuberculosis (Mtb) LeuRS [20]. The boron atom in the molecules was critical for Mtb LeuRS activity since it forms a bidentate covalent adduct with the terminal nucleotide of tRNA, Ade76, and traps the 3′ end of tRNALeu in the editing site to inhibit leucylation and thus protein synthesis. The (S)-aminomethyl side chain at C-3 in 4 and 5 was critical for binding, and C-4 halogen atoms (Cl and Br) significantly improved Mtb LeuRS activity, antitubercular activity against Mtb H37Rv, and selectivity against other bacteria. However, potential toxicity issues from the inhibition of mammalian cytoplasmic LeuRS and tolerance issues of 5 in a once daily dose of 50 mg/kg in a mouse acute tuberculosis (TB) infection model caused serious concerns for its use as a TB drug in long-term therapy. Throughout a series of medicinal chemistry campaigns to examine the structure–activity relationship (SAR) of 3-aminomethylbenzoxaborole derivatives, compounds 6–10 were found to improve both selectivity against human cytoplasmic LeuRS and HepG2 cell toxicity compared with 4 and 5, presumably by decreased lipophilicity and increased polarity in the molecule (Figure 4) [21]. Triaging lead compounds through in vivo pharmacokinetic (PK) analysis and efficacy assays using acute and chronic mouse TB infection models identified 8 (GSK656) as a preclinical candidate for tuberculosis; 8 successfully completed its first-time-in-human (FTIH) study to evaluate the safety, tolerability, pharmacokinetics, and food effects of single (5, 15, and 25 mg) and repeat oral doses (5 and 15 mg, 14 days, qd [quaque die, once a day]; NCT03075410) [22]. Moreover, 8 is currently in a phase 2 clinical trial for drug-sensitive pulmonary tuberculosis to evaluate its early bacterial activity, safety, and tolerability (NCT03557281). Details of the clinical studies of 8 will be discussed in a later section.
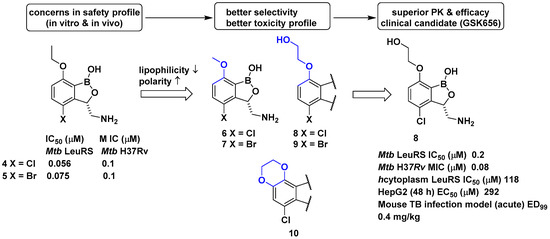
Figure 4. Development of GSK656 through hit-to-lead optimization.
Cladosporin isolated from Cladosporium cladosporioides and Aspergillus flavus was found to have potent antimalarial activity against both liver- and blood-stage Plasmodium falciparum by targeting the parasite cytosolic lysyl-tRNA synthetase (Pf LysRS) and terminating protein biosynthesis [23]. Despite its high clearance and poor oral bioavailability in vivo [24][25], the favorable selectivity and potency shown by cladosporin against Pf LysRS over human LysRS spurred medicinal chemistry efforts to further develop it to overcome its metabolic limitations as an oral drug. In this regard, Rusch et al. analyzed cladosporin by incubating it with mouse liver microsomes in vitro and revealed that three major metabolically weak points might cause the metabolic liabilities of cladosporin (Figure 5) [26]. The hydroxyl group, tetrahydropyran, and C-4 of isochromenone were potentially the three major metabolically weak points. Additionally, the X-ray crystal structure of cladosporin and P. falciparum LysRS (Pf LysRS) suggested an H-bond with Glu332 and the π/cation ‘cage' from adjacent aromatic and charged residues, and the interaction between the lactone ring and Arg559 through water as the major interactions [27]. Based on these basic findings, medicinal chemistry efforts were focused on the three metabolically weak points of cladosporin to generate the three major derivatives A (12), B (13), and C (14) as representative examples. Upon screening these libraries using the Transcreener AMP assay system to assess aminoacylation by LysRS [23], the unsaturation of the isocoumarin core and transformation of the metabolically unstable tetrahydropyran ring to the cyclohexyl ring yielded a selective LysRS inhibitor with improved metabolic stability at the level of hepatic extraction. In line with a previous report by Rusch et al. [26] focusing on metabolic weak points of cladosporin, Zhou et al. confirmed that the methyl tetrahydropyran moiety of cladosporin could be replaced by a more stable methylcyclohexane, while maintaining potency in the ATP hydrolysis assay (compounds 15 and 16) [28]. The methyl group in the cyclohexane ring was important for the hydrophobic interaction with Ser344, resulting in increased potency of 4-fold compared to that of compound 15. The replacement with a lactam group or a conjugated Δ3,4 double bond within the isocoumarin ring was tolerated, but the two phenolic hydroxyl groups were critical for binding to LysRS (Figure 5).
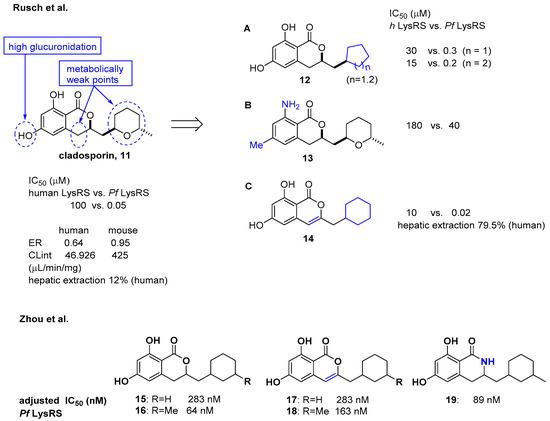
Figure 5. Optimization of cladosporin to improve pharmacokinetic properties [26][28].
Another medicinal chemistry campaign to develop a new antimalaria and anti-cryptosporidiosis drug targeting Pf LysRS1 and Cryptosporidium parvum (Cp) LysRS was continued by Baragaña et al. using cladosporin as a starting point [29]. Its low druggability due to high metabolic instability was optimized by replacing the isocoumarin core with the chromone scaffold connected to a metabolically stabilized cyclohexane ring with an amide linker, as shown in Figure 6. Compound 21 was active against both Pf LysRS1 (IC50 = 0.015 μM, luciferase ATP consumption assay) and whole-cell bloodstream P. falciparum 3D7 (EC50 [the half maximal effective concentration] = 0.27 μM) and was selective compared with human (Hs) LysRS (IC50 = 1.8 μM). The pharmacokinetic properties were optimized by stabilizing the metabolically labile cyclohexyl ring and blocking the point of potential hydroxylation of the phenyl group in the chromone core. As a result, compound 21 showed good systemic exposure with excellent oral bioavailability (F = 100%) and a moderate half-life (t1/2 = 2.5 h). In vivo efficacy was evaluated using a NODscidIL2Rγnull mouse (SCID) model and doses up to 40 mg/kg (po [per os, oral administration]), and it was found that a daily oral dose of ED90 = 1.5 mg/kg (1.0–2.3 mg/kg) (estimated daily exposure in blood AUCED90 [area under the curve at ED90] = 11,000 ng·h/mL/d (6900–14,000 ng·h/mL/d)) reduced parasitemia by 90%. A high level of sequence identity within the active site region of Pf LysRS1 and Cp LysRS (96% identity) and an overall sequence identity of 47.7% and similarity of 64.6% across the entire protein was observed from the evaluation of compound 21 against Cryptosporidium parvum both in vitro and in vivo. Compounds for cryptosporidiosis treatment must also have good exposure in the gastrointestinal tract and possibly also some systemic exposure since the parasite is found predominantly in the gastrointestinal tract with some in the biliary tract. In this regard, compound 21 was worthy of consideration for cryptosporidiosis since 17% of the oral dose was found in mouse stools, suggesting that some biliary excretion had occurred. Compound 21 showed efficacy in vivo in two different Cryptosporidium mouse models, NOD SCID gamma and INF-γ-knockout, at 20 mg/kg (po). However, the toxicity concern of 21 at 50 mg/kg in mice (po) at which the blood concentration in mice reached the EC50 for HepG2 cells limited further development. It is presumed that the toxicity is due to the inhibition of mammalian LysRS.
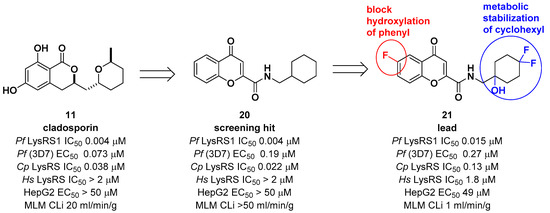
Figure 6. Optimization of cladosporin by replacing the core with a chromone scaffold.
Considering the significant structural differences between cladosporin derivatives (11–19) and the newly found chromone derivatives (20 and 21), Baragaña et al. analyzed the binding modes of cladosporin and 20 by studying their X-ray co-crystal structure with Pf LysRS1 (Figure 7) [29]. Cladosporin binds to Pf LysRS1 within the ATP-binding pocket with the isocoumarin moiety occupying the same space as the adenine ring of ATP, and the tetrahydropyran system taking the same position as the ribose ring of ATP. The two phenolic hydroxy groups of cladosporin form hydrogen bonds with E332 and the NH of N339, while the carbonyl interacts with a highly coordinated conserved water molecule. Although 20 binds with Pf LysRS1 in the ATP-binding pocket in a similar manner to cladosporin, the bicyclic core is rotated 30° anticlockwise with respect to cladosporin. The chromone core stacks between F342, H338, and R559. The ring carbonyl forms an H-bond to the backbone NH of N339, mimicking the N1 of adenine and the O1 OH of cladosporin. The amide carbonyl H bonds to a highly conserved water molecule coordinated by the side chain of D558 and R559. The cyclohexyl moiety projects into a pocket formed by the side chains of R330, F342, and S344 and the backbone of L555 and G556. This pocket is completed by the substrate lysine and is similar to that occupied by the tetrahydropyran ring of cladosporin, except that the cyclohexyl ring probes deeper into the pocket [29].
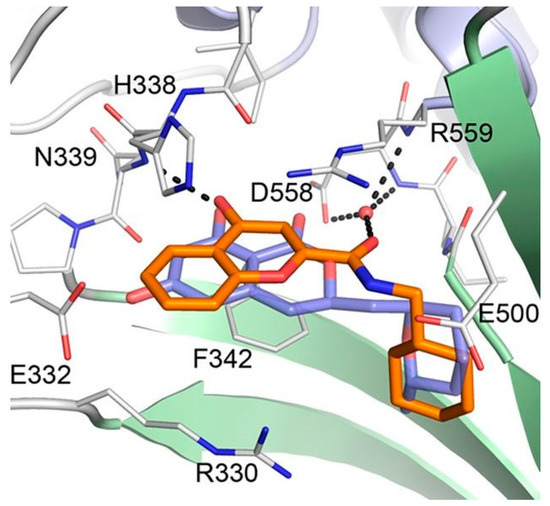
Figure 7. Binding modes of cladosporin and 20 to Pf LysRS1. Pf LysRS1:Lys:20 showing the binding mode of 20 (C atoms, gold) bound to the ATP site of Pf LysRS1 (PDB ID code 6AGT) superimposed upon Pf LysRS1:Lys:cladosporin (cladosporin C atoms, slate; PDB ID code 4PG3). The image was reproduced from reference 29 by Baragaña et al. and adapted for repurposing.
Borrelidin is known as a potent inhibitor of threonyl-tRNA synthetase (ThrRS) for malaria. However, its lack of selectivity against human ThrRS and high toxicity to human cells limit further development as a drug candidate. In this regard, Novoa et al. derived a series of borrelidin analogs that circumvent these potential problems by showing higher selectivity and lower cytotoxicity from minor structural changes in the carboxylic acid side chain [30]. In particular, compound BC196 (23), which released rotational constraints by adopting linear carboxylic acid, and compound BC220 (24), which converted terminal carboxylic acid to pyridyl ethyl ester on the cylcopentane ring, showed improved selectivity against P. falciparum over human HEK293 cells by 13,579-and 4048-fold, respectively. Considering that borrelidin exhibited 355-fold activity difference between P. falciparum and human HEK293 cells, selectivity was improved by 11–38-fold. From in vivo efficacy studies using a Plasmodium yoelii 17XL-infected mouse model, 23 and 24 showed 100% survival over 20 days after drug treatment at 6 mg/kg per day. In particular, 24 showed complete clearance of parasites in the blood at 6 mg/kg per day, 80% survival, and ~3% parasitemia at 0.25 mg/kg per day. It is also interesting that 24 was not cleaved to borrelidin by esterase, as determined in time course experiments with infected red blood cells (RBCs) over 27 h, suggesting that the biological effect of 24 is not due to its conversion to borrelidin by RBCs or plasmodial esterases (Figure 8).
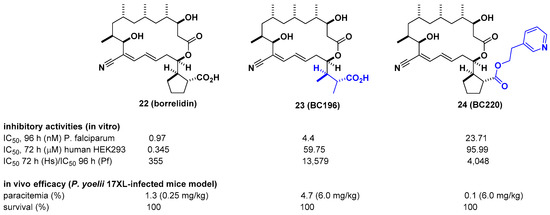
Figure 8. Optimization of borrelidin selectivity for Plasmodium falciparum over human threonyl-tRNA synthetase (ThrRS).
Unlike other LysRS inhibitors that target the active site of pathogen-derived LysRS, Kim et al. reported a new class of small molecule inhibitors, including YH16899, that bind to human LysRS without affecting the catalytic activity of LysRS [31]. Physiologically, it is known that LysRS binds to the 67-kDa laminin receptor (67LR), a target for antimetastatic therapeutics [32], in the plasma membrane and enhances cell migration that causes cancer metastasis. Thus, it is thought that directly inhibiting the interaction between LysRS and 67LR by a small molecule could suppress cancer metastasis and derive a new class of antimetastatic cancer therapy. In this regard, 25 showed specific inhibition of the binding between the LysRS and 67LR pair based on in vitro pull-down and immunoprecipitation (IP) assays and decreased the amount of membranous 67LR using LysRS-overexpressing H226 squamous lung carcinoma cells (Figure 9). The in vivo efficacy of 25 was demonstrated using three different in vivo mouse models, including (i) a mouse breast cancer model (4T1 cells, 100 mpk [mg per kilogram] and 300 mpk, po, ~60% inhibition of tumor metastasis), (ii) a Tg (MMTV-PyVT) model (100 mpk, po, ~70% reduction of pulmonary nodule formation), and (iii) a cancer cell colonization model (A549 cells expressing red fluorescence protein (RFP), ~50% reduction of tumor metastasis in the brain and bone tumors over 7 weeks). Overall, Kim et al. showed that the protein–protein interaction of LysRS-67LR is a promising approach for the development of new therapeutics for human diseases.
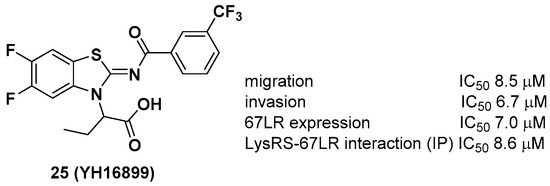
Figure 9. YH16899, a protein–protein interaction inhibitor of LysRS and 67-kDa laminin receptor (67LR).
Halofuginone (HF, 26), a well-known antifibrotic agent in preclinical and clinical studies, inhibits prolyl-tRNA synthetase (ProRS) activity [10]. Mechanistically, 26 competitively binds to the proline-binding pocket of the catalytic site of ProRS and causes drug resistance due to the accumulation of proline [33]. It is anticipated that the antifibrotic effect of 26 would be diminished in fibrotic tissue because a higher concentration of proline is often observed in fibrotic tissue than in nonfibrotic tissue [34]. In this regard, Shibata et al. from Takeda Pharmaceuticals developed T-3833261 (27), a potent ATP competitive inhibitor of ProRS that does not bind to the proline-binding site [11]. The antifibrotic activity of 27 was evaluated both in vitro and in vivo. From the transforming growth factor beta (TGF-β)-induced fibrotic assay, 27 inhibited the expression of fibrotic markers, including α-smooth muscle actin (α-SMA) and type I collagen (Col1a), by reducing mothers against decapentaplegic homolog 3 (Smad3) expression in human skin fibroblasts. These results were directly correlated with the in vivo TGF-β-induced mouse model. The topical application of 27 reduced the expression of fibrotic genes such as Col1a1, Col1a2, and α-SMA (Figure 10).
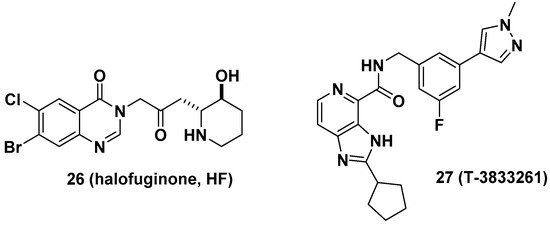
Figure 10. Antifibrotic prolyl-tRNA synthetase (ProRS) inhibitors halofuginone and T-3833261.
In a related study, it was presumed that the proline-rich nature of profibrotic proteins such as collagens may be attributed to glutamyl-prolyl-tRNA synthetase (GluProRS)-mediated translational regulation during cardiac fibrosis. Several studies with indirect evidence support the regulatory role of GluProRS as a target gene for cardiac fibrosis. For example, hypoactive mutations in the ProRS domain of GluProRS lead to hypomyelinating leukodystrophy without causing any known cardiac dysfunction in patients, implying that reduced ProRS enzymatic activity may not adversely affect normal heart function while reducing proline-rich protein synthesis [35]. In this regard, Akashi Therapeutics used 26 to treat Duchenne muscular dystrophy (DMD), analogous to reducing fibrosis by 26 and increasing muscle strength [36][37][38][39]. Recently, Wu et al. examined GluProRS-mediated regulatory mechanisms of profibrotic protein synthesis at the translational level and identified increased GluProRS in failing human and mouse hearts [40]. Increased GluProRS contributed to the elevated translation of proline (Pro)-rich (PRR) mRNAs, and genetic knockout of the glutamyl-prolyl-tRNA synthetase gene (Eprs) reduced fibrosis in different heart failure mouse models. Additionally, the selective GluProRS inhibitor 26 reduced the translation of PRR mRNAs, such as collagens and other novel PRR genes, including latent TGF-β-binding protein 2 (LTBP2) and sulfatase 1 (SULF1).
Aminoacyl-tRNA synthetase-interacting multifunctional protein 2 (AIMP2), a scaffold in the multi-tRNA synthetase complex (MSC), dissociates from the MSC and influences the activity of the p53, TGF-β, tumor necrosis factor-α (TNF-α), and Wingless/int-1 (WNT) signaling pathways [41][42]. A splicing variant of AIMP2 lacking exon 2 (AIMP2-DX2) correlates positively with cancer by compromising the tumor-suppressive function of native AIMP2 through competitive interactions with p53 fuse-binding protein (FBP) and TNF receptor-associated factor 2 (TRAF2) [43]. Lim et al. showed that AIMP2-DX2 binds with heat shock protein 70 (HSP70) and is stabilized by blocking the seven in absentia homolog 1 (Siah1)-dependent ubiquitination of AIMP2-DX2, which leads to cancer progression in vivo [44]. Representative AIMP2-DX2 inhibitors (28~30) are described in Figure 11. Based on this mode of action of AIMP2-DX2, the protein–protein interaction inhibitor BC-DXI-495 (28) was found to specifically inhibit the AIMP2-DX2 + HSP70 interaction and suppress cancer cell growth in vitro (IC50 = 4.2 μM, luciferase assay; EC50 = 14.2 μM, cell viability assay, A549 cells) and AIMP2-DX2-induced tumor growth in vivo (~50% decrease, 50 mpk, ip [intraperitoneal injection], 2 weeks, H460 stably expressing AIMP2-DX2 WT [wild type], BALB/cSLC mice). Consecutively, Sivaraman et al. reported BC-DXI-843 (29), which was optimized from BC-DXI-495 [45]; 29 significantly improved in vitro activity in luciferase and cell viability assays (IC50 = 0.92 μM, luciferase assay; EC50 = 1.2 μM, cell viability assay, A549 cells). The in vivo efficacy of 29 in the H460 xenograft mouse model was comparable with that of 28 by inhibiting ~60% tumor growth (50 mpk, bid [bis in die, twice a day], ip, 2 weeks). In the same context, Lee et al. separately developed the 2-aminophenylpyrimidine derivative 30 for AIMP2-DX2 interaction inhibition with >80% tumor growth inhibition in an H460 xenograft mouse model [46].
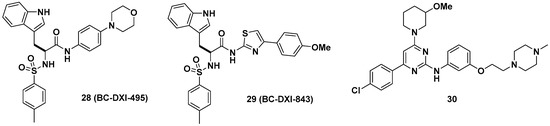
Figure 11. Aminoacyl-tRNA synthetase-interacting multifunctional protein 2 lacking exon 2 (AIMP2-DX2) interaction inhibitors.
Lastly, Kato et al. reported bicyclic azetidines such as 31 and 32 (Figure 12) as potent inhibitors of Pf phenylalanyl-tRNA synthetase (PheRS) (EC50 = 9 and 5 nM, respectively, against a multi-drug resistant strain, Pf Dd2) for the treatment of malaria [47]. Lead optimization campaign allowed the improvement of in vitro potencies of 31 against Plasmodium cynomolgi (EC50 = 3.34/2.86 μM for small form [SF, hypnozoite-like]/large form [LF, schizont], respectively), P. falciparum liver-stage (EC50 = 1.31 μM), and P. falciparum transmission-stage, gametocyte IV–V (EC50 = 663 nM) to EC50 = 0.933/1.04 μM (for SF/LF), 0.34 μM, and 0.16 μM in 32, respectively. Issues in 31 such as poor solubility (<1 μM in PBS), high clearance in human and mouse liver microsomes (Clint = 142 and 248 μL/min/mg, respectively), and a high volume of distribution in vivo (12 L/kg) were optimized by replacing the hydroxymethyl of 31 to N,N-dimethylaminomethyl group in 32 to give improved solubility (15 μM in PBS), decreased clearance in human and mouse liver microsomes (Clint = 31 and 21 μL/min/mg, respectively), and a better volume of distribution in vivo (24 L/kg), resulting in a longer half-life (32 h) which can be suitable for single-dose oral treatment. In vivo efficacy studies of 32 in multiple stages of malaria models including Plasmodium berghei CD-1 and P. falciparum huRBC NSG mouse models showed complete elimination of blood- and liver-stage parasites by a single dose during the study period. This study has served as an example of developing a cure for malaria as well as the potential to give prophylaxis and prevent disease transmission. Additionally, Vinayak et al. reported that bicyclic azetidine 32 is a potent inhibitor of Cryptosporidium parvum PheRS (Cp PheRS) for diarrheal disease in young children [48]. The in vivo efficacy of 32 for cryptosporidiosis was shown in immunosuppressed mouse models. Overall, it is thought that bicyclic azetidines such as 32 possess a unique core skeleton and warrant further examination to exploit their chemical space for applications not only in infectious diseases, but also for other diseases.
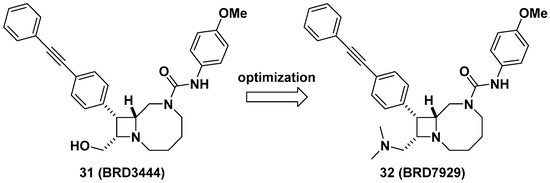
Figure 12. BRD7929, a potent inhibitor of Pf PheRS for malaria.





