1000/1000
Hot
Most Recent
 +1 point
+1 point
(Trans)membrane enzymes are typically not considered as good catalyst in biotechnology, due to difficultlies in production and purification. However, a growing number of applications are proposed in bioelectrocatalysis as membrane enzymes catalyse a number of energy-conversion reactions that are key in society. Bioelectrocatalytic systems that have been developed broadly fall in two catagories: (a) enzymatic biofuel cells and (b) biophotoelectrocatalyst. In order for these systems to function efficiently, the interface between the electrode surface and the membrane enzymes needs to be specifically tailored to retain the structural integrity of these amphiphilic macromolecules while - at the same time - enable fast electron exchange between the solid surface and the protein. This entry summarises key approaches in this area, which we here coin as "membrane protein modified electrodes".
Membrane proteins constitute 20–30% of all proteins encoded by both prokaryotic and eukaryotic cells. They perform a wide variety of functions, including material transport, signal transduction, catalysis, proton and electron transport (Figure 1) [1]. They are also key to a number of earth’s most fundamental reactions, such as respiration and photosynthesis [2][3]. Redox enzymes in the respiratory chain catalyse a variety of fundamental processes for energy conversion and fuel production, including H2 oxidation, O2 reduction, and carbon and nitrogen cycling. Membrane proteins that are involved in the light reaction of photosynthesis harvest light and facilitate electron transfer essential for solar energy conversion. The amphiphilic nature of membrane proteins makes them difficult to isolate, study and manipulate. Despite these challenges, membrane proteins have been widely advocated and studied for applications in bioelectrocatalysis, such as biofuel cells [4] and semi-artificial photosynthesis [5]. Here, we will review electrochemical studies of membrane proteins with the view to using these systems for bioelectrocatalysis. To aid discussion later on, we will briefly introduce a small selection of membrane enzymes active in bioenergy conversion, although this is far from a comprehensive list. We will then summarise the main strategies to immobilise membrane proteins on electrodes and discuss common techniques used to characterise these electrodes, including electrochemistry, spectroscopy, spectroelectrochemistry, microscopy and quartz crystal microbalance. Finally, some critical application challenges and potential future research directions will be highlighted that might find application in bioelectrocatalysis. Specifically, we will focus on two emerging directions. One is the reconstitution of membrane proteins into hybrid vesicles to extend their functional lifetime. The other is the use of microorganisms for microbial electrosynthesis and semi-artificial photosynthesis.
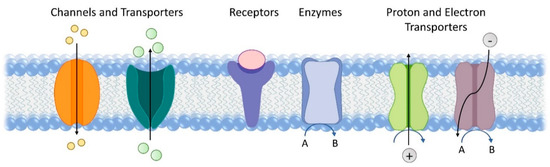
Figure 1. Representative functions of transmembrane proteins.
The increased demand to produce energy and value-added chemicals from cheap and environmentally friendly renewable resources has driven the recent advances in bioelectrocatalysis research towards the development of alternative systems. The applications of bioelectrocatalysis range from biosensors, energy conversion devices and bioelectrosynthesis [6][7][8][9]. The majority of the studies employ soluble proteins; however, some membrane proteins with suitable catalytic or electron transfer properties also find applications in bioelectrocatalysis. Some of them show advantages over soluble proteins or have unique functions. Here, we will discuss the membrane protein electrode applications in the field of bioelectrocatalysis.
One of the most studied energy conversion devices is the enzymatic biofuel cell (EBC) which uses oxidoreductase enzymes as biocatalysts to convert chemical energy into electrical energy [10][11][12]. A typical EBC consists of a two-electrode cell in which the biofuels (such as H2, formate) are oxidised at the bioanode and the oxidants are reduced at the biocathode (usually O2 is reduced to water) [13]. Among these, H2/O2 biofuel cells are one of the most investigated enzymatic systems [14]. Hydrogenases are promising biocatalysts to fabricate high performance H2-oxidation bioanodes. However, the extreme oxygen sensitivity of highly active hydrogenases is one of the main limitations of hydrogenase based H2 bioanodes [15]. The O2 sensitive hydrogenase can be protected by a low-potential viologen redox polymer matrix [16] or enzymatic O2 scavenger [17]. However, these protection mechanisms either consume the electron from H2 oxidation or add chemicals for protection. O2-tolerant MBH therefore shows great advantage for H2/O2 biofuel cell. The first membrane-less H2/O2 cell was assembled by Armstrong group. Two pyrolytic graphite electrodes, coated respectively with MBH from R. eutropha and laccase from Trametes versicolor (Tv), were immersed in a H2/air flushed solution. The system reached an OCV of ~970 mV and a maximum power output of ~5 µW [18]. High OCVs were achieved because MBH directly exchanged electrons with the electrode and no mediators were required. The Re MBH was solubilised from the membrane extracts by 2% Triton-X 114 and isolated via a Strep-tag sequence on the small subunit. It is possible that the MBH was devoid of the transmembrane cytochrome b anchor and this could explain why there was no need to use mediators. The Re MBH also showed CO-tolerance [18]. The same group further improved the performance of the H2/O2 cell by replacing the Re MBH with MBH (similarly Strep-tagged on the small subunit) from R. metallidurans, which is more active and stable to O2 exposure (Figure 2a). The authors emphasised that such a system would have the advantage to perform H2 conversion even in H2-poor mixtures. They also showed that three cells in series provided a total OCV of 2.7 V which was sufficient to power a wristwatch for 24 h [19].
As described earlier, we have presented a strategy for using the full heterotrimeric MBH as a biocatalyst (including the cytochrome b anchor) and have used multi-layer membrane stacks on gold electrode to increase MBH loading [20]. In our approach, we used cytoplasmic membrane extracts of R. eutropha and created interconnected layers of membranes with each layer containing anchored MBHs. Lipophilic quinones were used as mediator, shuttling electron between electrode and protein. However, the irreversible electrochemical behaviour of the quinone redox reaction increases the required overpotential for H2 oxidation, which will limit the power output of the devices and more research is needed to resolve this.
To optimise EBCs, gas diffusion electrodes have been investigated and enzyme coverage optimised. The porous structure of the gas diffusion electrode can increase the mass loading of the biocatalyst and overcome the mass transport limitation of gases. In 2016, Kano group used an O2-tolerant MBH from Hydrogenovibrio marinus and an O2-sensitive [NiFe]-hydrogenase from Desulfovibrio vulgaris “Miyazaki F” to create DET-type gas diffusion electrodes [21]. The authors did not specifically comment on whether their enzyme purification methods might have affected the presence of the cytochrome b anchor subunits of MBH. The MBHs were isolated through two different procedures. The large and small subunits of the O2-tolerant MBH were isolated through detergent solubilisation and maintained in 0.025% Triton-X. The O2-sensitive MBH went through a trypsinisation process which could lead to the separation of the transmembrane cytochrome b anchor. These H2 oxidation electrodes generated a current density of 10 mA∙cm−2 in the half-cell configuration. Contrary to the O2-tolerant MBH, the O2-sensitive MBH did not show overpotential for H2 oxidation and, on this basis, was selected by the authors as bioanode of a H2/O2 EBC for a further study. Coupling this O2 sensitive MBH anode with bilirubin oxidase (BOD) from Myrothecium verrucaria immobilised on Ketjen black-modified waterproof carbon papers (KB/WPCC) electrode, a dual gas-diffusion membrane- and mediator-less H2/air-breathing biofuel cell was constructed (Figure 3b) which showed maximum power density in the range of 6.1 mW∙cm−2 at 0.72 V [22].
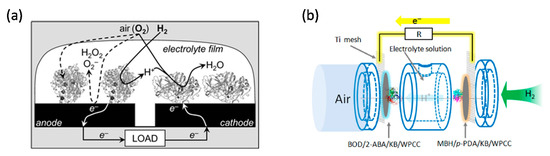
Figure 2. Schematic representations of enzymatic biofuel cells (EBC). (a) EBC comprises of graphite electrodes modified with O2-tolerant MBH of R. metallidurans CH34 (anode) and fungal laccase (cathode) in aqueous electrolyte exposed to 3% H2 in air. Reprinted with the permission from ref [20]. Copyright (2006), The Royal Society of Chemistry. (b) A dual gas diffusion membrane-free H2/air powered EBC comprises of a [NiFe]-MBH (anode) and a bilirubin oxidase (cathode). Reprinted with the permission from ref [22]. Copyright (2016), Elsevier B.V.
The O2-reduction biocathodes for EBC are usually based on multi-copper oxidases like bilirubin oxidase or laccase which can reduce O2 almost without overpotential [23]. Among the membrane proteins, cytochrome c oxidase has been studied as O2 reduction catalyst. Katz and Willner assembled a membrane-less glucose/O2 biofuel cell with cytochrome c/cytochrome c oxidase as O2 reducing cathode [24]. The cytochrome c was assembled on a maleimide modified gold electrode through a cysteine residue to link cytochrome c oxidase. In a follow-up work, the authors developed an electroswitchable and tunable biofuel cell. In this case, the cathode was modified with poly(acrylic acid) loaded with Cu2+ to covalently attach cytochrome c and link the latter to cytochrome c oxidase [25]. Although cytochrome c was able mediated electron transfer to cytochrome c oxidase for O2 reduction, only an OCV of 0.12 V was obtained, likely limited by the redox potential of cytochrome c (~0.25 V vs. SHE), which is much lower than that of H2O/O2 (0.82 V vs. SHE at pH 7). Although cytochrome c oxidase is not commonly used for O2 reducing cathode in biofuel cell, the recent work with gold NPs by the group of Hellwig has shown renewed possibilities for cytochrome c oxidase as catalyst. Recently, Wang et al. [26] reported that cytochrome c oxidase from acidophilic bacterium Acidithiobacillus ferrooxidans can reduce O2 at exceptionally high electrode potentials (+700 to +540 mV vs. NHE). The low overpotential for O2 reduction of this cytochrome c oxidase makes it an attractive biocatalyst as cathode of biofuel cells in the future.
Besides the use of H2 as fuel for EBC, formate is also a valuable feedstock for biofuel cells because its redox potential is similar to H2/H+. The Kano group reported a mediated electron transfer type formate/O2 biofuel cell by coupling formate dehydrogenase modified bioanode with BOD modified biocathode [27]. In nature, some formate dehydrogenases can catalyse the inverse reaction to reduce CO2 to formate [28]. However, to the best of our knowledge, the research in this field has been conducted only on soluble formate dehydrogenases. The ability of CO2 reduction and the applications of membrane-bound formate dehydrogenase need to be further explored.
Respiratory nitrate reductase (Nar) catalyses NO3− reduction to NO2−, this is an essential step to produce NH3. Today there is no enzyme known to reduce NO3− to NH3 directly [29]. DET with nitrate reductase has been shown to support the electrochemical reduction of NO3− to NO2− [30][31]. A full reduction of NO3− to NH3 was demonstrated by a cascade electrocatalysis process which combined nitrate reductase and noble metal catalyst to reduce NO2− to NH3 [32].
Biophotoelectrodes fabricated on planar carbon or SAM-modified metal electrode usually show low photocurrent density which limits their applications in bioelectrocatalysis. However, the development of redox polymer electrodes, layer-by-layer assembly and 3D architectures has enhanced the performance of biophotoelectrodes [33][34]. PSII is the only natural protein able to catalyse the photooxidation of water. The electron flow can be blocked by herbicide compounds since they bind to the terminal plastoquinone QB of PSII. This inhibition effect can be exploited for designing PSII light-driven biosensor to detect herbicides [35]. A similar approach to detect herbicides was taken with purple bacteria RCs [36].
As water oxidation biophotocatalyst, PSII attracts more attention for solar energy conversion to generate electricity or fuels. As mentioned above, PSII photoanodes have been connected to PSI photocathodes (both in Os redox polymers) to mimic natural photosynthesis Z-scheme for solar-to-electricity generation [37][38]. In a similar approach using a benzoquinone redox polymer, a PSII photoanode (O2 evolution) has been connected to a bilirubin oxidase cathode for O2 reduction [39]. Recently, PSII was integrated together with PbS quantum dots within a TiO2 inverse opal electrode to perform H2O photooxidation. This was combined with an inverse opal antimony-doped tin oxide (ATO) cathode modified with bilirubin oxidase to catalyse oxygen reduction. This system achieved a high open-circuit voltage of about 1V under illumination (Figure 3a,b) [40].
A full water splitting process can be realised by combining a PSII photoanode with a cathode modified with hydrogenases. In contrast to EBCs, water-soluble hydrogenases (typically [FeFe] hydrogenases, but also [NiFe] hydrogenases; H2ases) are most commonly used for water splitting systems. When combining PSII and H2ases, there is an energy gap between the terminal electron acceptor QB within PSII and one of the FeS cluster within H2ase. A biophotoelectrochemical cell with a PSII photoanode and a H2ase cathode thus requires an applied bias voltage of 0.8 V to drive H2O splitting [41]. This was improved by wiring H2ase and p-Si on an inverse opal TiO2 photocathode, lowering the required applied bias voltage to 0.4 V for H2O splitting [42]. Finally, by integrating PSII on a diketopyrrolopyrrole dye-sensitised TiO2 photoanode and connecting it with a H2ase cathode, a bias-free photoelectrochemical cell for H2O splitting was developed by the group of Reisner [43]. Coupling a dye-sensitised PSII photoanode with a W-dependent formate dehydrogenase (FDH) cathode, a biophotoelectrochemical cell was constructed for CO2 reduction at a small bias voltage of 0.3 V (Figure 3c,d) [44]. The latter study showed the possibility for rational design of biophotoelectrochemical cells for value-added chemicals generation beyond H2.
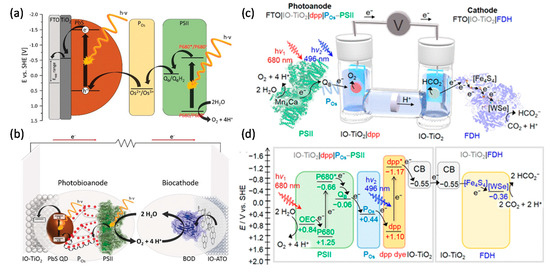
Figure 3. Photoelectrochemical (PEC) cell for electricity or biofuels generation. (a) Schematic of the electron transfer steps and energetic level of the components of the light-driven signal chain composed of TiO2, PbS QDs, redox polymer (POs), and PSII. (b) A scheme of a PEC cell consisting of an IO-TiO2|PbS|POs|PSII anode and an IO-ATO|PC|BOD cathode. Reprinted with the permission from ref [181]. Copyright (2019), Wiley. (c) A schematic representation of a semi-artificial photosynthetic tandem PEC cell coupling CO2 reduction to water oxidation. A blend of POs and PSII adsorbed on a dpp-sensitized photoanode (IO-TiO2|dpp|POs-PSII) is wired to an IO-TiO2|FDH cathode. (d) Energy level diagram showing the electron-transfer pathway between PSII, the redox polymer (POs), the dye (dpp), the conduction band (CB) of IO-TiO2 electrodes, four [Fe4S4] clusters, and the [WSe]-active site in FDH. All potentials are reported vs. SHE at pH 6.5. Reprinted with the permission from ref [44]. Copyright (2018), American Chemical Society.
Unlike PSII, PSI does not directly catalyse technologically valuable reactions such as water oxidation. However, photoexcitation of PSI provides the reductive potential to drive reactions with other catalysts, such as Pt and H2ase [45]. Recent studies show that it is possible to drive H2 production from light with PSI and H2ase by electrode design. Photoelectrodes were manufactured in a ‘layered’ fashion using an Os redox polymer, PSI and, finally, a polymer/H2ase mix [46]. Photoelectrochemical H2 production is achieved at an onset potential of +0.38 V vs. SHE. In this study, the PSI was randomly orientated and did not form a compact layer, likely limiting efficiency by charge recombination between the carrier and mediator or electrode. In a more recent study, an anisotropically oriented PSI monolayer was formed using Langmuir-Blodgett deposition [47]. A compact and oriented PSI layer minimises charge recombination and enables unidirectional electron transfer to H2ase for H2 evolution. Combining this PSI/H2ase photocathode with a PSII photoanode created a system able of bias-free light-driven water splitting [47]. Langmuir-Blodgett deposition transfers only a monolayer of PSI and this might limit the performance of the biophotoelectrode as this limits the loading or coverage of PSI.
One of the limitations with biophotoelectrodes is the limited lifetime of the isolated proteins, especially PSII [7]. Light-induced formation of reactive oxygen species can further limit the lifespan of proteins [48]. Another limitation of biophotoelectrodes is that photosynthetic proteins only use a limited range of the solar spectrum which reduces solar conversion efficiency. The absorption spectral range can be enhanced by attaching complementary chromophores to light-harvesting complex proteins [49]. It also can be improved by integrating biological light-harvesting antenna complexes or organic dyes/synthetic compounds to the RCs [50][51].





