1000/1000
Hot
Most Recent
 +1 point
+1 point
Mitophagy is a form of macroautophagy, were mitochondria and their contents are ubiquitinated, engulfed, and removed through lysosome degradation.
Mitochondria are essential organelles that regulate energy homeostasis, cell signaling, and cell death [1][2][3]. During threatened cell death or nutrient starvation, mitochondria can be degraded and recycled through mitophagy. Mitophagy is a specific form of autophagy, a process where cell contents are degraded and recycled. In a broad sense, mitophagy involves tagging mitochondria for removal, engulfment of the organelle by an autophagosome, and degradation in a lysosome. There are several pathways which control, initiate, and facilitate mitophagy. The many facets of mitochondrial function contribute to mitophagy pathways.
Mitochondria coordinate and balance energy production through beta oxidation, the citric acid cycle (TCA cycle), and oxidative phosphorylation at the electron transport chain (ETC). Beta oxidation is a catabolic pathway where free fatty acids are converted to acetyl coA, which enter the TCA cycle and ultimately oxidative phosphorylation. In the TCA cycle, either pyruvate (from glycolysis) or acetyl coA are oxidized to generate the high energy electron carriers, NADH and FADH2. NADH and FADH2 enter the ETC at complex I and complex II, respectively. These high energy electron carriers undergo oxidation/reduction reactions in the ETC in order to pump protons into the matrix. These protons ultimately power ATP synthase (or Complex V) for the generation of ATP from ADP. These bioenergetic reactions maintain the mitochondrial electrochemical gradient, or mitochondrial membrane potential. Mitochondrial membrane potential is the main signal which either inhibits or initiates mitophagy.
Mitochondria are double membrane organelles. The outer mitochondrial membrane is imperative for mitophagy function [4][5][6][7]. Transport and signaling proteins localize to the outer mitochondrial membrane to facilitate protein and metabolite import. As discussed in more detail below, aggregation-prone proteins are known to block these import channels on the outer mitochondrial membrane in some neurodegenerative diseases. This could lead to the disruption of mitophagy processes and requires more research efforts to understand. The inner membrane space of mitochondria houses enzymes and allows for proton storage and protein folding. The mitochondrial inner membrane contains the ETC and ATP synthase enzymes and the mitochondrial matrix stores enzymes for the TCA cycle, mitochondrial DNA (mtDNA), and other crucial enzymes for protein folding and maintenance of pH gradients. For more detailed analysis of mitochondrial localized proteins MitoCarta3.0 was recently published [8].
Synaptic loss is strongly correlated with cognitive deficits and motor dysfunction [9][10][11][12]. Mitochondria are essential for synaptic function and neurotransmitter synthesis, release, and uptake [13][14][15]. Accumulation of damaged mitochondria could lead to synaptic dysfunction and neurodegeneration. Mitophagy may play a role in ensuring synaptic mitochondrial integrity by degrading damaged mitochondria.
Mitochondria evolved from a prokaryotic endosymbiont. As such, mitochondria share characteristics with bacteria including a double membrane, circular DNA, formyl-methionine amino acids, and cardiolipin [1]. In certain contexts failure of mitophagy pathways could lead to the release of mitochondrial components into the extracellular space, activation of a damage-associated molecular response (DAMP), and inflammation [1][5]. Mitochondria are also master regulators of cell death pathways (such as apoptosis and necrosis) [4][5][16]. As a by-product of the respiratory chain function superoxide radicals are produced. These free radicals generate multiple species of reactive oxygen species (ROS) and reactive nitrogen species (RNS). During periods of mitochondrial dysfunction and failure of mitochondrial quality control mechanisms (such as mitophagy) ROS/RNS can induce damage to cellular macromolecules and necrotic cell death [4][5][16]. Proper control and coordination of mitophagy pathways are crucial to prevent cell death and inflammation.
Mitophagy is imperative for glial cell function. Signaling between microglia, astrocytes, and neurons are modulated by mitophagy pathways. Novel data show that transcellular mitophagy pathways occur within the brain. Transcellular mitophagy is a process by which cells release mitochondria for engulfment and mitophagy in surrounding cell types. Dysregulation of this process can lead to neuroinflammation and loss of proteostasis [17][18][19].
Disruption of mitophagy is observed with aging and in many neurodegenerative diseases. Recent advances have described novel mechanisms of mitophagy within the central nervous system (CNS). Here, we will review current knowledge of mitophagy regulation, its role in neurodegenerative disease, and therapeutic potential.
For this review article, we used PubMed, clinicaltrials.gov, and Google Scholar to identify studies related to mitophagy, AD, PD, ALS, and MS. We used search terms including mitophagy, mitophagy and neurodegeneration, mitophagy and AD, mitophagy and PD, mitophagy and MS, mitophagy and ALS, mitochondria and neurodegeneration, and autophagy.
Autophagosomes can be derived from membranes of endoplasmic reticulum (ER), Golgi, mitochondria, or plasma membrane [20][21][22][23][24]. The biogenesis of autophagosomes involves the formation of the isolation membrane (IM), elongation and maturation, closure, and then fusion with the lysosome to form the autolysosome. The first step in autophagosome biogenesis is the activation of the Unc-51-like kinase 1 complex (ULK1; pre-initiation complex), which contains ULK1, autophagy-related proteins 13 and 101 (Atg13, Atg101), and focal adhesion kinase family interacting partner 200 (FIP200) [25][26][27][28]. This complex recruits the class III phosphatidylinositide 3-kinase (PI3K) Vps34 complex (Beclin1, autophagy-related protein 14 (Atg14), autophagy and beclin 1 regulator 1 (Ambra1), and vascular protein sorting 34 and 15 (Vps34 and Vps15)) to produce phosphatidylinositol 3-phosphate (PI3P), also called the initiation complex [25][27][29][30]. PI3P binding proteins, FYVE domain containing proteins (DFCP1), and WD repeat protein interacting with phosphoinositide (WIPIs) localize to the IM [26]. All of this culminates in the formation of the omegasome and IM. Autophagy-related proteins 12, 5, and 16 (Atg12, Atg5, and Atg16) and LC3-phosphatidylethanolamine (PE) facilitate the elongation and closure of the IM, following fusion with the lysosome [25][26][31].
Mitochondrial quality control mechanisms include mitochondrial chaperones, the mitochondrial unfolded protein response (UPRmt), degradation of mitochondrial proteins in the cytoplasm via the proteasome (p97, 26S proteasome), removal of damaged proteins via mitochondrial derived vesicles (MDVs), and mitophagy (Figure 1) [32]. Misfolded mitochondrial proteins can be refolded by mitochondrial chaperone proteins (heat shock proteins 22, 60, and 70) or cleaved/degraded by mitochondrial proteases Lon and Clp through the UPRmt [33][34][35][36][37]. Damaged mitochondrial proteins can also be targeted specifically for proteasome degradation by p97 through the proteasome [38]. MDVs bud off mitochondria after engulfing damaged mitochondrial macromolecules. MDVs are associated with impaired mitochondrial import channels. These MDVs are either degraded by lysosomes, peroxisomes, or exocytosed [39][40]. Mitophagy (reviewed extensively below) functions to remove damaged mitochondria with the goal of preventing cell death.
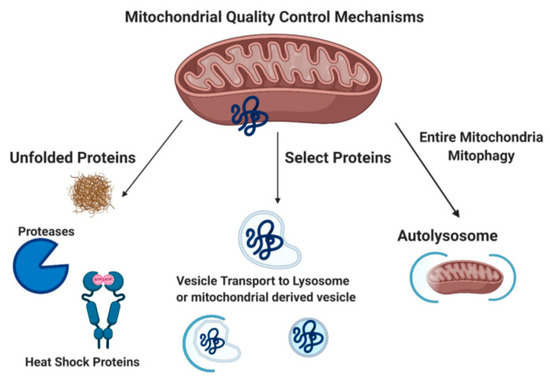
Figure 1. Mitochondrial Quality Control Mechanisms. Unfolded protein response, mitochondrial derived vesicles, and mitophagy [23][33] Created with BioRender.com.
Increasing mitophagy in transgenic mouse models of neurodegeneration have shown mostly beneficial effects. In AD models (iPSC derived, transgenic mice, and C. elegans), increasing mitophagy using nicotinamide mononucleotide (NMN), UA, or actinonin (AC) reduced Aβ and tau aggregation. In AD transgenic mice, mitophagy induction benefited cognition [41][42]. These compounds are NAD+ precursors, which may drive mitophagy through alterations in redox balance (NAD+/NADH). UA likely drives mitophagy through a PINK1/Parkin/Nix axis.
Broad autophagy induction with Rilmenidine in the G93ASOD1 mouse model of ALS did not change disease progression [43]. The mechanism(s) of Rilmenidine autophagy/mitophagy induction are currently unknown. Rapamaycin (an mTOR inhibitor) treatment of this same mouse model was detrimental unless mature lymphocytes were depleted [44]. These studies highlight the importance of understanding the non-cell autonomous effects of autophagy and mitophagy pathways.
In PD rodent models (MPTP injection), a drug, Salidroside, increased Parkin and PINK1 expression and preserved dopaminergic neurons in the substantia nigra [45]. A cell permeable form of Parkin rescued cells from aggregating α-synuclein, partially restored motor function, and protected dopaminergic neurons in the 6-OHDA PD (6-hydroxydopamine) mouse model [46].
In an MS-related mouse model of EAE administration of rapamycin, an mTOR inhibitor improves outcomes [47]. Further studies of the EAE mouse model show that excessive activation of Drp1 through nitration leads to an overaction of mitophagy [48]. Blocking this pathway alleviated the disease burden in the EAE mouse mode [49]. Genetic ablation of Beclin 1 was also protective in the EAE mouse model [50]. Overall, in MS inhibition of mitophagy specifically in T cells could be beneficial.
UA has been shown to be safe and well-tolerated in elderly adults, with plasma concentrations detectable at a range of doses. Furthermore, UA affected mitochondrial gene expression in muscle [51]. A separate study in healthy adults is registered for UA (NCT04160312), but no results have been posted. Clinical trials for NMN (NCT04228640 safety trial) are recruiting or ongoing (NCT03151239 effects on cardiometabolic health). In Japan, the first human clinical trial of NMN showed no deleterious effects, suggesting NMN is tolerable and safe [52][53]. No clinical trials for these NAD+ precursor mitophagy modulators are currently registered for neurodegenerative diseases.
Lifestyle interventions could be useful tools to boost mitophagy. Exercise and diet have been shown to induce mitophagy [54][55][56][57][58]. In both AD animal models and human clinical trials, exercise has shown cognitive benefit [57][59][60][61][62][63]. The exercise effects in ALS and PD are more controversial, but overall, exercise seems to improve physical and cognitive outcomes [64][65][66][67][68][69]. Intermittent fasting and ketogenic diets have also been shown to induce mitophagy and improve cognition/motor performance [70][71][72][73][74][75][76].
Current clinical trials aimed at increasing autophagy, mitophagy, or mitochondrial function are ongoing or recently completed. For AD, these include treatment with nicotinamide riboside (NR; NCT04430517; NAD+ precursor), Dimebon (NCT00675623, NCT00829374; stimulates mTOR-dependent mitophagy), resveratrol (NCT00678431; mTOR inhibitory), ketogenic diets (NCT03860792), and caloric restriction diets (NCT02460783). In PD, these include nicotinamide supplementation (NCT03568968; NAD+ precursor), ubiquinol/Coenzyme Q10 (NCT03061513; autophagy mechanism unknown), ketogenic diets, and ketone esters (NCT01364545, NCT04477161). In MS, clinical trials include ketogenic diet, dimethyl fumarate, and MitoQ (NCT03740295, NCT04267926, NCT02461069). In ALS, one clinical trial for ubiquinol/Coenzyme Q10 (NCT00243932; autophagy mechanism unknown) was completed. Overall, the clinical trials directly modulating mitophagy are lacking and require more attention. The majority of mitophagy inducers in clinical trials have unknown mechanisms and pleotropic affects.
Targeting specific pathways and tissues could be advantageous in avoiding deleterious or off-target effects. Designing new therapeutic strategies should focus on modulating specific mitophagy targets while also enhancing mitochondrial function and biogenesis.





