1000/1000
Hot
Most Recent
 +1 point
+1 point
Amyotrophic lateral sclerosis (ALS) is the most frequent motor neuron disease and a neurodegenerative disorder, affecting the upper and/or lower motor neurons. Notably, it invariably leads to death within a few years of onset. Although most ALS cases are sporadic, familial amyotrophic lateral sclerosis (fALS) forms 10% of the cases. In 1993, the first causative gene (SOD1) of fALS was identified. With rapid advances in genetics, over fifty potentially causative or disease-modifying genes have been found in ALS so far. Accordingly, routine diagnostic tests should encompass the oldest and most frequently mutated ALS genes as well as several new important genetic variants in ALS.
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by both upper and lower motor neuron degeneration, paralysis, and ultimately limiting survival from two to five years after onset [1]. Disease onset typically occurs in late middle-life with the mean age being 65 years. It results in relentless progressive muscle atrophy and weakness, ending with respiratory failure [2][3]. In addition, this neurodegenerative disorder has an estimated worldwide mortality rate of 30,000 patients per year [4]. ALS cases are estimated to occur in 2–3 per 100,000 individuals in Europe, and less than one in Asia [5], thus categorizing it as a rare disease. Furthermore, up to 10% of ALS-affected individuals have an affected family member or members with familial ALS (fALS), in which most have inherited the disease in an autosomal dominant manner [6]. The remaining ALS patients, with no clear genetic linkage, are called sporadic ALS (sALS) [7]. At present, mutations in over 50 genes have been shown to contribute to the ALS pathogenesis [8][9]. Some of them such as SOD1, C9orf72, FUS, and TARDBP were shown to present deleterious mutations, while other variants mostly found by association studies rarely occur in the less frequent genes [8][9][10]. Several studies have identified oxidative stress, glutamate excitotoxicity, apoptosis, neurofilament dysfunction, protein misfolding and aggregation, impairment of RNA processing, disrupted axonal transport, endosomal trafficking dysfunction, inflammation, and mitochondrial impairment as the molecular pathways which lead to the disease and indicate ALS pathogenesis. RNA metabolism and protein metabolism have been indicated as commonly proposed ALS pathogenic mechanisms [11].
Since the end of the 19th century, there have been many studies on ALS, but its pathogenesis is still unclear [4]. Many genetic variants that contribute to the missing heritability of ALS are yet to be discovered. Nowadays, the number of studies on finding novel ALS genes and their functions on disease pathogenesis is growing quickly [12][13][14][15][16][17][18][19][20]. This review discusses the current genetic understanding and landscape of ALS and summarizes its disease mechanisms. Here, we specifically review the genes involved in ALS in three categories, the most prevalent, the less frequent (≤1%), and the most recent ALS genes, and depict how genomic signatures turn into pathogenic molecules in ALS.
To date, 5–10% of all ALS cases suffer the known mutant genes [21][22]. SOD1 is the most frequently mutant gene with 20% prevalence; mutant TARDBP and FUS in 5% and 4% of fALS cases, respectively, have been identified as well. In the United States and Europe, 40% of fALS cases suffer C9orf72 variants, and the rest are caused by variants in other known or unknown genes (Figure 1) [23]. Simultaneously, variants in C9orf72 and SOD1 genes, which occur most frequently, constitute 7% and 1% of sALS cases, respectively [24]. To provide historical context, we illustrate the yearly number of publications from PubMed that include the terms “ALS” and “the gene name” in the title and abstract (Figure 1C). According to the timeline of gene discoveries, SOD1, TARDBP, FUS, and C9orf72 are the first to fourth genes found with more than 1% occurrences in the patients in the years 1993, 2006, 2009, and 2011, respectively. Furthermore, there are some genes involved in ALS with less frequent variants (≤1%) and several recent genes over the past two years with relatively rare variants such as CYLD, S1R, GLT8D1, and KIF5A, which are precisely discussed in this review.
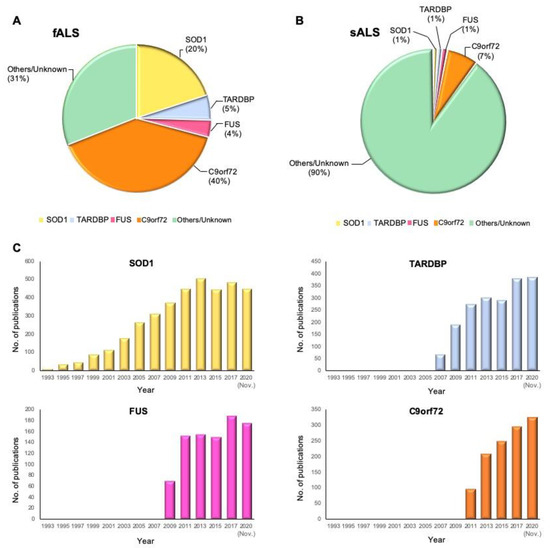
Figure 1. Proportional contribution of mutated genes in ALS and timeline of ALS gene study. (A) The proportion of mutant genes in fALS cases. (B) The proportion of mutant genes in sALS cases. (C) Timeline of ALS gene discoveries and researches for SOD1, TARDBP, FUS, and C9orf72. The Y-axis shows the number of publications in PubMed that include the terms “ALS” and “the gene name” by year until November 2020.
Several novel ALS-associated genes have been proposed by the researchers over the past two years. Farhan et al. identified DNAJC7 as a novel gene for ALS that encodes a member of the heat-shock protein family, HSP70, and has a key role in protein function such as protein folding and stabilization. Alteration of HSP70 and DNAJC7 gene expressions causes protein aggregation in ALS model [17]. Another gene, WDR7, was proposed by Course et al., in which the human-specific 69 bp variable number tandem repeat in the last intron of this gene may be associated with ALS. WDR7 repeat expansions may act similar to the specific range of CAG repeat expansion numbers at ATXN2, which are enriched in ALS cases. It was shown that WDR7 repeat expansion could form microRNAs, RNA aggregates, and lead to RNA editing [18]. Further study has shown that ATXN1 overexpression disturbs TDP-43 nucleocytoplasmic transport, which leads to a decrease in the nucleocytoplasmic ratio of TDP-43. Hence, mislocalization and aggregation of TDP-43 can be considered the hallmark of ALS [19]. Finally, ACSL5, a neurotoxic A1 astrocyte-related gene, is upregulated in ALS cases. ACSL5 can induce A1 astrocytes, leading to motor neuron death and ALS progression. Overexpression of ACSL5, similar to the previously discovered gene GPX3, is associated with rapid weight loss in humans [8][20]. Although the mentioned genes above have been identified as ALS associated genes, the study on their contribution to ALS pathogenesis is still limited.
Dobson-Stone et al. identified a novel missense variant in CYLD gene as the genetic cause of ALS in a large European Australian family [16]. Previously, they found a disease locus on chromosome 16 by genome-wide linkage analysis [25]. The missense variant in CYLD leads to alteration of CYLD immunoreactivity in the brain tissue [16], and shows two ALS-associated pathological phenotypes: an elevation of cytoplasmic TDP-43 level [26] and an impairment of autophagy function [27]. Overexpression of CYLD inhibited transport of TDP-43 into the nucleus from the cytoplasm. In the nucleus, TDP-43 controls the expression of ATG7, which mediates the fusion of lysosomes with autophagosomes. Decreased expression of ATG7 resulted in a loss of autolysosome formation [26][27][28]. Notably, overexpression of CYLD leads to the failure of autophagosome–lysosome fusion, causing the malfunction of autophagy [16][29] (Figure 2A).
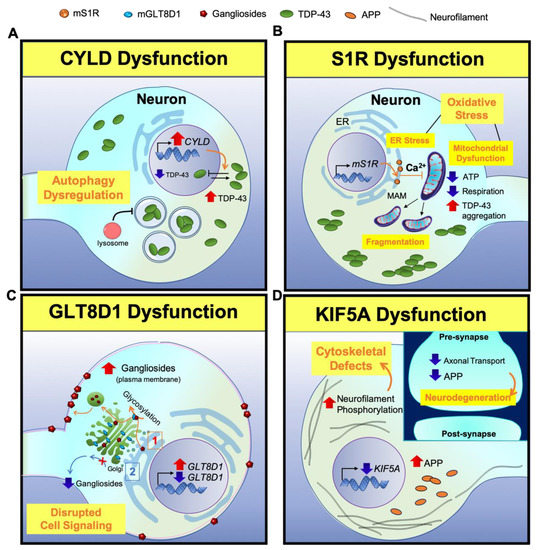
Figure 2. A scheme for illustrating neuropathogenesis of CYLD, S1R, GLT8D1, and KIF5A genes in ALS. (A) CYLD: Overexpression of CYLD inhibits the nuclear transport of TDP-43 from the cytoplasm, leading to the mislocalization of TDP-43. Lower TDP-43 level in the nucleus decreases the expression of ATG7, which is responsible for the fusion of lysosomes with autophagosomes. This results in a loss of autolysosome formation, causing a loss of autophagy function. (B) S1R: Mutant S1R leads to ER calcium and ATP depletion and perturbs mitochondrial dynamics and respiration. Subsequently, depletion of ATP leads to TDP-43-induced toxicity. (C) GLT8D1: (1) Mutant GLT8D1 increases the ganglioside signaling, leading to the transit of mature gangliosides to the cell membrane where they disrupt cell signaling. (2) On the other hand, knock down of GLT8D1 impairs its glycosyltransferase activity from the Golgi and diminishes ganglioside signaling. (D) KIF5A: Lack of KIF5A expression disrupts axonal transport, accumulates phosphorylated neurofilaments and APP, and causes cytoskeletal defects in neurons. (Inlet) In contrast, the disruption of axonal transport reduces APP level in the synapse and triggers neurodegeneration.
SOD1, TARDBP, FUS, and C9orf72, as the oldest and most frequently mutant ALS genes, are described and their function in ALS pathogenesis are illustrated (Figure 3).
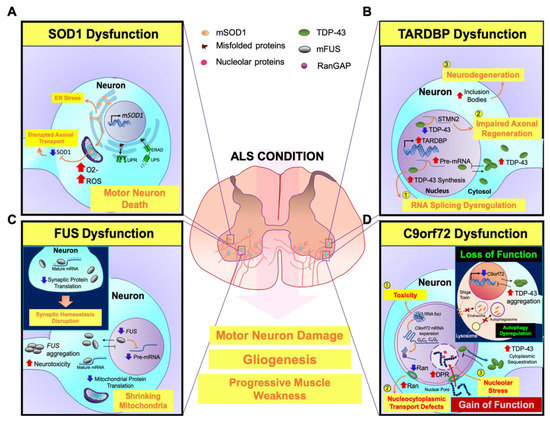
Figure 3. A scheme for illustrating neuropathogenesis of mutant genes (SOD1, TARDBP, FUS, and C9orf72) with major contribution to ALS. (A) SOD1: Mutant SOD1 (mSOD1) leads to oxidative stress by elevating free radicals that damage protein folding and impair axonal transport in motor neurons. Additionally, misfolded mSOD1 protein is not able to transport across mitochondrial membranes and accumulates onto the outer mitochondrial membrane, triggering motor neuronal death. On the other hand, mSOD1-induced ER stress results in the activation of UPR and ERAD, causing motor neuronal damage. (B) TARDBP: (1) Mutant TDP-43 (mTDP-43) is aggregated in the cytosol and TDP-43 dysfunction abnormally accumulates mRNA in the nucleus. (2) The loss of nuclear TDP-43 function leads to inhibition of TDP-43 binding to STMN2 RNA and mis-splicing of STMN2 RNA, which subsequently inhibits axonal regeneration of motor neurons. (3) On the other hand, upregulation of cytoplasmic TDP-43 forms inclusion bodies and it further propagates to neighbor cells, causing motor neuronal degeneration. (C) FUS: Mutant FUS (mFUS) disrupts nucleocytoplasmic transport and leads to its depletion from the nucleus. Then, mFUS aggregates in the cytoplasm and induces neurotoxicity. mFUS may also disrupt translation of transcripts associated with mitochondrial function, which leads to mitochondrial shrinkage. (Inlet) Moreover, mFUS binds to mature mRNAs in the cytoplasm and affects mRNA translation. Suppressed protein translation impairs the dendrites and axon terminals. Inhibition of local intra-axonal protein translation, that are required for synaptic maintenance and function, causes synaptic defects and dysfunctions. (D) C9orf72: (1) The nuclear RNA foci are generated by the aggregation of repeat-containing C9orf72 RNAs in the nucleus and cause neurotoxicity. (2) Sequestration of RanGAP by G4C2 RNA disrupts the nucleocytoplasmic transport function. The loss of nuclear Ran depletes nuclear TDP-43 levels and elevates cytoplasmic TDP-43 levels. (3) Furthermore, the imported dipeptide repeats (DPRs) into the nucleus are associated with nucleolar proteins and cause nucleolar stress. (Inlet) The loss of C9orf72 function in endosomal trafficking regulation by interactions with nuclear pore complex proteins eventually increases cytoplasmic TDP-43 inclusions. Furthermore, reduction of C9orf72 expression inhibits Shiga toxin transportation from the plasma membrane to the Golgi apparatus, and alters the ratio of LC3, an autophagosome marker, leading to autophagy dysregulation.
Nowadays, by developing next-generation sequencing technologies, rare genetic variants were discovered in several studies. However, the specific link between gene variants and disease mechanisms is sometimes unclear because of the small number of patients carrying those variants. Functions of the discovered genes can be categorized into four categories: (1) genes that influence RNA processing such as ANG [30], SETX [31], MATR3 [32], ATXN2 [33], TAF15 [34], EWSR1 [35][36], ELP3 [37], hnRNPa1, hnRNPA2B1 [38], ATXN1 [39], and GLE1 [40]; (2) genes involved in protein trafficking and degradation such as ALS2 [41], VAPB [42], CHMP2B [43], FIG4 [44], UBQLN2 [45], SQSTM1 [46], SIGMAR1 [47], OPTN [21], VCP [48] and TBK1 [49]; (3) genes that influence cytoskeletal and axonal dynamics such as DCTN1 [50], PFN1 [51], SPG11 [52], TUBA4A [53], NEFH [54], PRPH [55], NEK1 [56], C21orf2 [57]; and (4) CHCHD10 [58] and C19orf12 [59] involved in mitochondrial dysfunction.
Although we have come a long way since proposing SOD1 as the first ALS gene 27 years ago, the causative pathogenic mechanisms in ALS remain obscure. The function of mutant CYLD, S1R, GLT8D1, and KIF5A as the recently discovered genes involved in ALS pathogenesis is illustrated in Figure 2. In addition, Figure 3 provides previous findings of the genetic effects of mutant SOD1, TARDBP, FUS, and C9orf72 and the fundamental mechanisms of ALS pathogenesis. According to the genes and genetic functions reviewed in this paper, multiple factors are involved in ALS disease development and progression, in which the most commonly proposed ALS pathogenic mechanisms are RNA metabolism and protein metabolism. Furthermore, genetic and phenotypic variants between patients make it difficult to draw general conclusions on ALS pathogenesis and predict the future outcomes in ALS research. However, further research on finding novel genes, gene modifiers, and their molecular pathways might improve our understanding about this neurodegenerative disorder, which is still fatal. In addition, new therapeutic strategies, by either the identification of shared disease pathways or targeted therapies known for genetic variants can be developed by proposing novel ALS causal genes. As of now, riluzole is the only successfully established therapy for ALS that exerts transient effects on cortical and axonal hyperexcitability [60], whereas several drug trials targeting glutamatergic neurotransmission have been unsuccessful [61]. Regarding the irreversibility of genetic variants, developing therapeutic approaches is difficult and specific drugs that target the treatment of ALS are quite limited [62]. Considering how genetic variants result in epigenetic modifications and how epigenetic alterations are reversibly modulated in various neurodegenerative disorders such as Alzheimer’s disease and Huntington’s disease, it would be reasonable to design therapeutic approaches that target the epigenetic components to ameliorate the onset and symptoms of ALS [63][64][65]. Therefore, in future studies, a combination of treatments that modulate the multiple targets of epigenetic components could be the more effective therapeutic strategy for treating ALS.





