1000/1000
Hot
Most Recent
 +1 point
+1 point
The clinical efficacy of anti-epidermal growth factor receptor (EGFR) antibody cetuximab for oral squamous cell carcinomas (OSCCs) is low. We previously reported that an increased oncogenic ROS proto-oncogene 1 (ROS1) is responsible for the invasiveness and metastasis of OSCC. This study demonstrates for the first time that ROS1, a receptor tyrosine kinase, can localize to mitochondria. Mitochondrial ROS1 in the highly invasive OSCC promotes mitochondrial fission, enhances mitochondrial oxidative phosphorylation and ATP production but reduces mitochondrial biogenesis. These findings highlight the novel function of ROS1 in mitochondrial morphogenesis and metabolic adaptation to promote OSCC invasiveness.
Oral cancer is the sixth-most-common cancer worldwide, and approximately 90% of oral cancers are oral squamous cell carcinomas (OSCCs). Unfortunately, most patients with oral cancer are diagnosed at an advanced stage with neck lymph-node metastasis[1]. The monoclonal antibody therapeutic cetuximab, which targets epidermal growth factor receptor (EGFR), is the most commonly prescribed treatment for advanced OSCC[2], although its clinical efficacy is limited[3][4] owing to drug resistance or lack of response[5]. Therefore, understanding the molecular mechanisms underlying OSCC metastasis is essential for designing more effective therapeutic approaches.
Receptor tyrosine kinases (RTKs) are synthesized in the endoplasmic reticulum, delivered to the Golgi, and then targeted to the plasma membrane as single-transmembrane proteins where they transduce extracellular signals to orchestrate diverse physiological responses[6]. The ROS proto-oncogene 1 (ROS1) encodes an RTK containing a large N-terminal extracellular domain, a single-transmembrane domain, and an intracellular, C-terminal tyrosine kinase domain. We previously showed that upregulation of ROS1 oncogene leads to oral cancer metastasis[7]. The highly invasive OSCC lines, OC3-IV2 and C9-IV2, express increased levels of ROS1. These two cell lines were in-vivo selected, highly invasive cells[7][8]. In addition, genomic rearrangements in the ROS1 gene have been implicated in cancer progression [9]. Unlike ROS1 which has been assumed to localize at the plasma membrane [10], ROS1 fusion proteins derived from chromosomal translocations exhibit differential subcellular localizations and their oncogenic potential can vary. For example, in glioblastoma, the FIG-ROS1 oncoprotein containing a Golgi-targeting signal, targets to the Golgi rather than the plasma membrane resulting in oncogenic transformation[11]. Similarly, the endosome-localized fusion proteins SDC4-ROS1 and SLC34A2-ROS1 and endoplasmic reticulum-localized fusion protein CD74-ROS1 differentially activate mitogen-activated protein kinase (MAPK)[12]. These findings suggest that subcellular distribution of ROS1 may determine its oncogenic properties. To address this issue, we examined the subcellular localization of ROS1—the results provide new insight into the role of ROS1 in regulating OSCC invasiveness.
The dynamics of mitochondrial morphogenesis correlate with changes in bioenergetics, cell metabolism, and motility[13]. To determine the physiological implications of mitochondrial morphogenesis, we measured the mitochondrial oxygen consumption rate (OCR) and ATP synthesis in OC3 and OC3-IV2 cells. The fraction of basal OCR inhibited by the ATP synthase inhibitor oligomycin was used to estimate the mitochondrial respiration rate required to sustain cellular ATP production. The protonophoric uncoupler carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) stimulates the mitochondrial electron transport system to function at its maximal capacity, defined as maximal OCR. The spare respiratory capacity is the difference between the basal and maximal OCR in response to a stress. Antimycin A and rotenone inhibit the mitochondrial electron transport system through Complexes I and III to allow measurement of nonmitochondrial respiration processes. The basal OCR was not significantly different between OC3 and OC3-IV2 cells. OC3-IV2 cells, on the other hand, exhibited a 55% enhancement of maximal OCR, a 2-fold elevation in spare respiratory capacity, and a 30% increase in ATP production compared to OC3 cells (Figure 1A,B), reflecting higher energy production in OC3-IV2 cells. This result revealed a link between spare respiratory capacity, ATP production, and cell invasiveness. To examine whether ROS1 directly regulates mitochondrial bioenergetics, we knocked down ROS1 in OC3-IV2 cells and determined the effect on mitochondrial OCR. Compared to control OC3-IV2-Scr cells, knockdown of ROS1 reduced basal OCR, maximal OCR, spare respiratory capacity, and ATP production (Figure 1C,D). These results demonstrated that elevated ROS1 enhances mitochondrial bioenergetics.
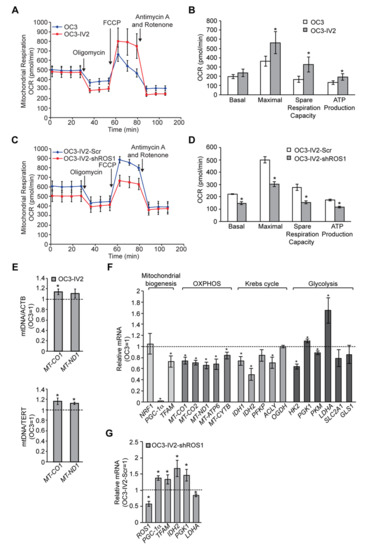
Figure 1. ROS1 affects mitochondrial function in OSCC cells. (A–D) Measurement of oxygen consumption rate (OCR) in OC3 and OC3-IV2 (A) or OC3-IV2-Scr and OC3-IV2-shROS1 cells (C) using the Seahorse XF24 analyzer. (B,D) Average basal OCR, maximal OCR, spare respiration capacity, and ATP production in OC3 and OC3-IV2 (B) or OC3-IV2-Scr and OC3-IV2-shROS1 cells (D) are shown. (E) The relative mtDNA content was measured by Q-PCR. The expression of MT-CO1 or MT-ND1 was normalized to nuclear-encoded ACTB or TERT from OC3 and OC3-IV2 cells. (F) Q-PCR of the mRNA levels of the indicated genes in OC3-IV2 cells normalized to that of OC3 cells. Genes were grouped into functional clusters related to mitochondrial biogenesis, OXPHOS, Krebs cycle, and glycolysis. (G) The mRNA levels of the indicated genes in OC3-IV2-shROS1 cells were normalized to those measured in OC3-IV2-Scr. Data from three independent experiments are presented as mean ± SEM (* p < 0.05, paired Student′s t-test).
It is possible that ROS1-dependent enhancement of mitochondrial bioenergetics may be due to increased mitochondrial biogenesis. To study this issue, we measured the mitochondrial DNA content and expression of genes associated with mitochondrial oxidative phosphorylation (OXPHOS) and mitochondrial biogenesis. OC3-IV2 cells had higher mitochondrial DNA content, whereas the expression of mitochondrial protein-coding genes of the OXPHOS system, namely MT-CO1, MT-CO2, MT-ND1, MT-ATP6, and MT-CYTB, was reduced in OC3-IV2 cells compared to OC3 cells (Figure 1E,F). Expression of the transcriptional cofactor PGC-1α and the master transcription factor TFAM for biogenesis was also downregulated in OC3-IV2 cells (Figure 1F). These data indicated that OXPHOS and ATP production in highly invasive oral cancer cells are concomitant with lower mitochondrial biogenesis. In addition, PGC-1α and TFAM expressions increased when ROS1 was reduced (Figure 1G), implicating a direct role for ROS1 in modulating mitochondrial biogenesis.
As efficient mitochondrial OXPHOS has been implicated in cellular metabolism[13], we investigated the effects of the ROS1-induced increase in mitochondrial bioenergetics on expression of genes encoding metabolic enzymes. Compared to OC3 cells, we found that the expression of genes associated with the Krebs cycle and glycolysis, including IDH1, IDH2, ACLY, HK2, and PKM, was reduced in OC3-IV2 cells, whereas PGK and LDHA were upregulated in OC3-IV2 cells (Figure 1F). Lactate dehydrogenase A, encoded by LDHA, converts pyruvate to lactate and generates ATP—and this pathway has been shown to be an essential source of energy for cancer cells[14]. We also found that ROS1 knockdown led to marked downregulation of LDHA (Figure 1G), suggesting that ROS1 can modulate the expression of LDHA. This is consistent with the idea that cancer cells maximize ATP production by both glycolysis and OXPHOS, resulting in the utilization of different nutrients and an increase in metabolic plasticity to maintain tumor survival and metastasis during different stages of disease progression[15]. Together, our findings describe a new role for the ROS1 oncogene in enhancing mitochondrial bioenergetics and metabolic plasticity to promote OSCC invasiveness.





