1000/1000
Hot
Most Recent
 +1 point
+1 point
T-cell acute lymphoblastic leukemia (T-ALL), a T-cell neoplasia that mainly affects children, is still a medical challenge, especially for refractory patients for whom therapeutic options are scarce. Recent advances in targeted immunotherapies for other hematopoietic neoplasias have engendered unprecedented expectations for the successful treatment of T-ALL patients, with the challenge still pending on establishing protocols for clinical management of associated side effects.
This review provides a comprehensive update on the different immunotherapeutic strategies that are being currently applied to T-ALL. We highlight recent progress on the identification of new therapeutic targets showing promising preclinical results, and discuss current challenges and opportunities for developing novel safe and efficacious immunotherapies for T-ALL. Collectively, we conclude that current progress on: 1) the application of universal off-the-shelf CAR T cells that prevent fratricide; 2) the incorporation of suicide genes allowing for CAR T-cell elimination after tumor eradication and T-cell immunodeficiency reversion; and 3) the discovery of increasingly specific molecular targets proved critical for disease progression in preclinical models, has tilted the balance between risks and benefits towards the use of immunotherapy for relapse/refractory T-ALL. Still, avoidance of associated adverse effects demands further efforts for the identification of new unique T-ALL antigens absent on normal T cells that guarantee the safe and effective application of these strategies.
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematological disorder that results from the progressive accumulation of genomic alterations in T-cell precursors developing in the thymus. T-ALL is characterized by the infiltration of bone marrow by immature T-cell lymphoblasts, while immature T-cell tumors characterized by a thymic mass and limited bone marrow infiltration are instead diagnosed as T-cell lymphoblastic lymphoma (T-LBL). T-ALL was described as an independent disease in the 1970s, after finding thymus-associated markers expressed on the surface of leukemic cells from pediatric patients [1]. Its incidence is higher in children than in adults (up to 25% and 15% of newly diagnosed ALL cases, respectively) [2], and it is twice as prevalent in males as in females [3]. Patients with T-ALL are frequently classified as high-risk due to the unfavorable features of the disease that include high leukocyte count, hematopoietic failure, and medullar and extramedullar infiltration with high probability of affectation of the central nervous system (CNS), which represents a frequent site of relapse [4]. In 1995, the European Group for Immunological Characterization of Leukemias (EGIL) established a classification of different clinically relevant T-ALL subtypes based on the expression of cell surface markers corresponding to sequential intrathymic T-cell developmental stages [5]: pro-T, pre-T, cortical and mature T-ALL (Figure 1). However, in recent years, improved genomic and transcriptomic techniques have provided new insights into the characterization of the prevalent genetic lesions involved in T-ALL pathogenesis [6][7][8], which has proved more valuable for risk-stratification of patients at the time of diagnosis [9][10][11]. Detection of translocations of enhancers or promoters of T-cell receptor (TCR) genes to other chromosomal regions helped the identification of the first T-ALL oncogenes, including the transcription factors TAL1 [12], LYL1 [13] and TLX1/HOX11 [14]. Unique gene expression profiles were later found to be associated with leukemic arrest of thymocytes at different developmental stages [15], leading to the definition of new T-ALL subgroups characterized by the driver oncogenes or oncogene fusions (TAL/LMO, TLX1, TLX3, HOXA, and MYB genes), denoted as type A aberrations, present at diagnosis [16]. Other genetic alterations, denoted as type B, are recurrently detected in T-ALL patients and include point mutations, insertions and deletions (INDELs), and chromosomal gains or losses, which result in activation of the NOTCH1 T-cell fate specification pathway (NOTCH1/FBXW7) in more than 60% of T-ALL cases [17], activation of cytokine signaling pathways (IL7R, JAK1/3, FLT3, CKIT, PI3K/AKT/PTEN, ABL1, N/KRAS) and transcription factors (RUNX1, ETV6, BCL11B, WT1, TCF7, LEF1, CTNNB1, GATA3, IKZF1), inactivation of cell cycle inhibitors (CDKN2A/B, CDKN1B, CDKN1C, CCND3, RB), or deregulation of chromatin modifiers and remodeling factors (PHF6, CTCF, KDM6A, SETD2, KMT2A/2D/2C, DNMT3A, IDH1/2) (reviewed in [18][19][20][21][22]).
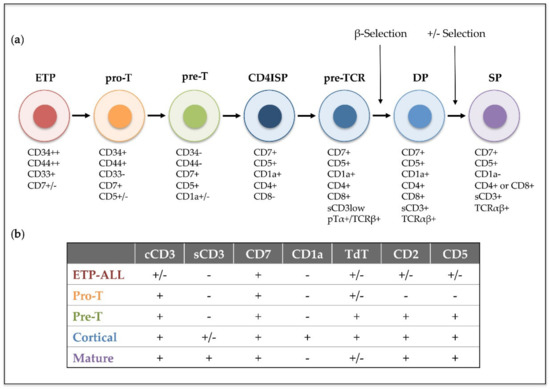
Figure 1. Human intrathymic T-cell developmental stages and corresponding T-ALL phenotypic subtypes. (a) Early thymic precursors (ETP) that have entered the human thymus differentiate through sequential developmental stages: pro-T, pre-T, immature CD4 single positive (CD4ISP), pre-T-cell receptor (pre-TCR), double positive CD4+ CD8+ (DP) and single posive (SP), which can be identified by the expression of different cell surface markers and the sequential acquisition of the pre-TCR and the TCRαβ receptors. (b) T-ALL subtypes (ETP-ALL, Pro-T, Pre-T, Cortical and Mature) are based on the 1995 EGIL classification of acute leukemias and the 2017 WHO classification of Tumours of Haematopoietic and Lymphoid Tissues, which included ETP-ALL as a new provisional entity. This classification was established by immunophenotyping based on surface molecules expressed at sequential intrathymic stages of human T-cell development. cCD3 (cytoplasmic CD3); sCD3 (surface CD3); TdT (terminal deoxynucleotidyl transferase). Presence or absence of the indicated molecule is represented by (+) and (−), respectively.
Nonetheless, molecular and genetic abnormalities were not considered for the characterization of T-ALL subtypes in the last revised edition of the World Health Organization (WHO) classification of ALLs in 2017 [23], and only early T-cell precursor lymphoblastic leukemia (ETP-ALL), which accounts for approximately 10% of pediatric and 40%–50% of adult T-ALL cases, was introduced as a new provisional entity [24]. ETP-ALL is characterized by the lack of T-lineage markers (preferentially CD1a, CD8 and weak CD5) but a phenotype that resembles that of early thymic progenitors (ETP), with expression of stem cell (CD34, CD117) and myeloid (CD13, CD33) lineage markers [25]. Genetically, ETP-ALL frequently associates with mutations in genes encoding epigenetic regulators (IDH1, IDH2, and DNMT3A), signaling factors (i.e., NRAS and FLT3), and transcription factors involved in hematopoietic and T-cell development (RUNX1, GATA3 and ETV6), whereas both activating NOTCH1 mutations and CDKN2A deletions co-occurring with oncogenic NOTCH1 mutations are rarely observed [26][27]. ETP-ALL has been for years associated with poor prognosis [25][28][29], but application of early response-based intensification regimens in the last years has greatly improved the outcome of these patients [30][31].
In the 1960s, only 20% of T-ALL patients were cured, but nowadays, intensive chemotherapy as the standard front-line therapy for T-ALL has raised cure rates to above 85%. Current protocols for T-ALL patients include consecutive phases of induction, consolidation, delayed intensification, and maintenance, with drug combinations that commonly include doxorubicin or daunorubicin, dexamethasone or prednisone, vincristine, asparaginase, cyclophosphamide and cytarabine, together with methotrexate and intrathecal chemotherapy as prophylaxis for CNS infiltration [32][33][34]. In a retrospective study, the Children’s Oncology Group (COG) reported that 5 yr overall survival (OS) for patients younger than 20 years who enrolled in their ALL clinical trials increased from 70.7% in 1990–1994 to 81.6% in 2000–2005 [35]. Similar 5 yr disease-free survival (DFS) and OS (83.8% and 89.5%, respectively) were obtained for all children and young adults (1 to 31 yr) enrolled in the AALL043 methotrexate early-intensification study by the same group from January 2007 to July 2014 [36]. However, adult T-ALL still presents a dismal outcome, with significantly lower survival rates than pediatric T-ALL. Although 90%–95% of adult patients achieved complete remission (CR) in different trials [37][38][39], OS after 3 and 5 years was only 65% and 48% respectively, with percentages decreasing with age to only 27% 5 yr OS for patients aged over 50 years. Relapse occurred in 30%–40% of adult T-ALL patients within the 7–24 months after remission and less than 10% of the relapsing patients survived [36][37]. Minimal residual disease (MRD) at the end of the induction phase is the key prognostic factor of relapse. MRD assessment in childhood T-ALL, either by real time quantitative polymerase chain reaction (PCR) detection of TCR gene rearrangements or by flow cytometry immunophenotyping of leukemic cells, has established MRD ≥ 10−3 as the most important predictive factor of relapse [40][41]. In adult T-ALL, MRD level ≥ 10−4 is associated with higher incidence of relapse and reduced OS, and has been a criteria used to classify high-risk patients [42][43].
The therapeutic available options for patients experiencing relapse or for those who are refractory to standard chemotherapeutic regimes are very scarce, and since the approval of nelarabine by the US Food and Drug Administration (FDA) in 2005 [44], no new agents have been specifically developed for T-ALL. This is certainly not the case for relapsed and/or refractory (r/r) B-cell acute lymphoblastic leukemia (B-ALL) patients, whose life expectancy has increased considerably in the last years after the introduction of anti-CD22 antibodies, bi-specific T-cell engagers (BITEs) and, lately, chimeric antigen receptors (CARs). Although nelarabine, a cytotoxic DNA damaging agent, has improved the survival of T-ALL relapsing patients [45][46][47], its dose-limiting toxicity [48][49][50], together with the absence of alternatives, underscore the need for new targeted therapies. However, the shared expression of surface markers between normal and leukemic T cells has limited the development of new targeted immunotherapies against T-cell malignancies and particularly, against T-ALL. This is due to the induction of secondary T-cell immunodeficiency is associated with therapy, which may result in the appearance of opportunistic infections and/or the reactivation of latent infections leading to life-threatening situations. Consequently, main challenges of future T-ALL treatments rely on (1) the identification of unique markers of T-ALL blasts, especially of those expressed on leukemia-initiating cells (LICs), which are the drivers of relapse [51], and (2) the elucidation of therapies aimed at killing leukemic but not healthy T cells, in order to avoid immunodeficiency.
We will also focus on novel molecules (Figure 2) emerging as promising T-ALL therapeutic targets in preclinical mouse models and discuss how their use is anticipated for clinical immunotherapy.
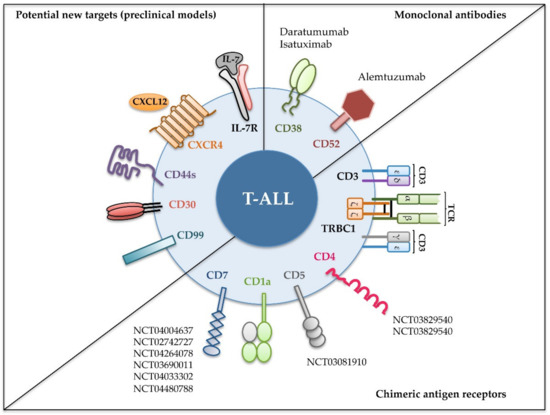
Figure 2. Recent immunotherapeutic interventions for T-ALL. Different strategies targeting the indicated cell surface molecules expressed on leukemic cells have been designed for T-ALL treatment. Monoclonal antibodies have been tested in the clinic against relapsed and/or refractory T-ALL (upper right); chimeric antigen receptors were assayed either in preclinical or in clinical settings (lower right); and new molecules have revealed their potential as promising targets in preclinical models (left). Identifiers [The National Clinical Trial (NCT) number] of clinical trials in progress including T-ALL patients are shown [52].
The identification of antigens with an expression restricted to malignant T cells has been the unsuccessful goal pursued by numerous investigations whose final aim is the development of suitable immunotherapy strategies that preserve the pool of healthy T cells. Development of powerful bioinformatics techniques in the last years has allowed the identification of few antigens that seem to be specific of leukemic T cells, not expressed by normal mature T cells nor by HSPCs [53][54]. Yet, the effectiveness of these molecules as immunotherapeutic targets remains to be investigated. Meanwhile, several surface proteins, despite being shared between leukemic T-cell, some normal T-cell subpopulations, and other non-T cell lineages, have emerged as critical players in T-ALL pathogenesis. Moreover, some of them, including CXCR4, CD44, and interleukin-7 receptor (IL-7R), have recently been validated as promising therapeutic targets in T-ALL preclinical animal models (Figure 2).
CXCR4 (C-X-C chemokine receptor type 4, CD184) is a seven-transmembrane domain G-protein-coupled receptor of the chemokine receptor family that interacts with CXCL12 (SDF-1). CXCR4 is expressed in several cell types including HSPCs, where it plays an important role in the maintenance of stem cell interactions with components of the bone marrow [55][56]. CXCR4 signaling is also essential for B lymphopoiesis [57][58] and for T-cell migration to different organs, including CNS [59][60][61][62][63].
The involvement of the CXCR4/CXCL12 signaling axis in the pathogenesis of several types of cancers was expected considering its implication in both cell migration and regulation of interactions with components of the tumor microenvironment, a function that may ultimately regulate metastasis [64][65]. The essential role of CXCR4 in T-ALL pathogenesis was recently demonstrated in a NOTCH1-induced T-ALL mouse model [66], where inhibition of endogenous CXCR4 expression by short hairpin RNA (shRNA) impaired CXCL12-induced migratory properties of T-ALL cells, and also induced cell death and altered cell cycle progression in vitro. When transplanted into immunodeficient mice, both murine and human CXCR4-silenced T-ALL samples showed delayed leukemia onset accompanied by increased host survival, as compared to control shRNA-transduced cells. This effect was mediated by reduced LIC frequencies and a deficient bone marrow homing of CXCR4-silenced T-ALLs. In vivo, T-ALL cells are found in close contact with bone marrow CXCL12-producing cells and this interaction is crucial for disease progression. In fact, CXCR4 targeting leads to remission of established murine T-ALLs and human PDXs [67]. In a different study, pharmacological CXCR4 antagonists inhibited bone marrow colonization by T-ALLs and reduced the neuropathologies associated with CNS infiltration [68], suggesting that CXCR4 may be a potential therapeutic target to eradicate CNS affectation in T-ALL patients. Several reports have suggested a relationship between Notch signaling and CXCR4 in T-ALL, although no direct regulation of CXCR4 expression by NOTCH has been documented. In a Notch1-induced T-ALL mouse model, bone marrow T-ALL cell colonization reduced the wild type HSPC pool, suppressed B-cell development, and caused thrombocytopenia, thus altering normal hematopoiesis [69]. These effects were mediated by a competition of T-ALL cells for perivascular regions and by the activation of Notch signaling in stromal cells, which led to a reduction in osteoblastic cells and a negative regulation of CXCL12 transcription. Therefore, T-ALL-mediated disruption of bone marrow niches promotes malignant progression at the expense of normal hematopoiesis through CXCR4/CXCL12 deregulation. Deregulated Notch3 activation also results in CXCR4 expression in preleukemic CD4+CD8+ intrathymic T cells that migrate to and colonize the bone marrow in a CXCR4-dependent manner [70].
While no differences in CXCR4 mRNA expression were detected between leukemic and normal T cells in microarray analysis [71], elevated CXCR4 surface expression was reported in T-ALL leukemic blasts compared to normal peripheral T cells, both in human and mouse [67]. This would be an advantage for implementation of CXCR4-targeted therapies. However, double-positive (DP) thymocytes express high levels of CXCR4, which raises important concerns about potential induction of thymic T-cell aplasia by CXCR4 inhibitors. Up to date, several small-molecule CXCR4 antagonists have been developed (reviewed in [72]), which mainly act by inhibiting CXCL12 binding and have been used for HSPCs mobilization from bone marrow [73][74]. Among them, Plerixafor (AMD3100) was approved by the FDA in 2008, in combination with G-CSF, for mobilization of HSPCs to peripheral blood prior to autologous stem cell transplantation. It has also been used for r/r AML [75][76] and CLL treatment showing promising results [77]. In a different trial, children with r/r AML, ALL or myelodysplastic syndrome (MDS) were treated with Plerixafor in combination with cytarabine and etoposide [78], and although AML patients achieved complete remission, no clinical response was observed in ALL and MDS patients. Another CXCR4 antagonist, POL5551, was shown to increase cytarabine sensitivity in a xenograft model of a high-risk pediatric ALL subtype, infant MLL-rearranged (MLL-R) ALL [79], indicating that disruption of the CXCR4/CXCL12 interaction may benefit children with high-risk ALL.
Based on the promising results obtained with pharmacological CXCR4 antagonists, several anti-CXCR4 antibodies have been produced and tested in preclinical models against different types of leukemia [80][81][82], and some of them have entered into clinical trials. Ulocuplumab (BMS-936564/MDX1338) was shown to block CXCL12 binding to CXCR4-expressing cells, inhibiting CXCL12-induced migration and inducing apoptosis in different hematological malignancies [83]. In a recent phase Ib/II study in adult patients with r/r MM, Ulocuplumab in combination with lenalidomide and dexamethasone resulted in a high response rate, although neutropenia and thrombocytopenia were observed in some patients [84]. Additionally, LY2624587 antibody was capable of inhibiting tumor growth in a CCRF-CEM T-ALL xenograft model by preventing CXCR4 signaling [85]. Therefore, given the potential antileukemic effects of CXCR4 inhibition in preclinical T-ALL assays, and the acceptable tolerance to different chemical inhibitors and antibodies already tested in patients, it may be worth expanding these studies on CXCR4/CXCL12-axis targeting to r/r T-ALL patients. Currently, there is an open phase IIa trial to assay the CXCR4 antagonist BL-8040 (NCT02763384) in combination with nelarabine in adult patients with r/r T-ALL/T-LBL.
IL-7R is a type I heterodimeric transmembrane receptor for IL-7, consisting of an α chain (IL-7Rα) and a common gamma chain (γc) that is shared between several cytokine receptors (IL-2, IL-4, IL-9, IL-15 and IL-21). IL-7Rα can also heterodimerize with the CRLF2 (cytokine receptor-like factor 2) subunit to generate a high affinity receptor for thymic stromal lymphopoietin (TSLPR) [86]. IL-7 was firstly described as a growth factor for developing B cells [87], but its role in cell survival and proliferation was quickly proved essential also for T cells [88][89]. IL-7-induced IL-7R signaling is indispensable to accomplish the full intrathymic maturation program of both gamma-delta (γδ) and alpha-beta (αβ) T cells [90][91][92][93]. It regulates the first expansion wave that bone marrow-derived progenitors seeding the thymus must undergo in order to generate the immature T-cell pool, which will afterward rearrange TCR genes and develop into either TCRγδ or TCRαβ mature T cells. IL-7 also plays a relevant role in the maintenance and survival of naïve [94][95] and memory peripheral T cells [96][97][98][99][100], as well as in innate lymphoid cell development [101][102]. Given that T-ALL arises from the malignant transformation of immature T-cell progenitors, it was not unforeseen that a high proportion (more than 70%) of T-ALL cases express functional IL-7Rs that mediate proliferative and prosurvival signals in response to IL-7 [103][104]. More recently, a possible role of IL-7R in T-ALL molecular pathogenesis started to become apparent after discovering that IL-7R is a direct target of NOTCH1 and is involved in Notch-mediated T-ALL cell maintenance [105][106][107]. Since the report in 2004 that NOTCH1 is a commonly mutated gene in T-ALL [17], deciphering the molecular mechanisms behind NOTCH1 deregulation leading to normal T-cell transformation, and elucidation of NOTCH1 target genes involved in leukemia cell growth, have been the keystones for developing novel targeted therapies for T-ALL patients harboring NOTCH1 mutations.
NOTCH1 signaling induces IL-7R expression by transcriptional upregulation of the IL7R gene (encoding IL-7R) through binding of the CSL/MAML-1 transcriptional complex to the IL7R promoter [107], a mechanism conserved in the mouse [106]. In addition, genome-wide mapping of NOTCH1 DNA-binding sites has highlighted a major role of super-enhancers in the dynamic regulation of NOTCH1 target genes including IL7R [108]. These findings indicate that the cell growth-promoting effects of the NOTCH1 oncogenic program are enhanced by means of IL7R gene induction and activation of IL-7R signaling, pointing to IL-7R downstream effectors as promising molecular targets for therapeutic intervention in T-ALL. As in normal T cells, IL-7R signaling in T-ALL cells is triggered by binding of IL-7 to, and heterodimerization of, IL-7Rα and γc chains, which places the intracellular domains of the receptor and the attached JAK1 and JAK3 kinases into close contact. This initiates serial phosphorylation events that activate STAT proteins (STAT1, STAT3 and STAT5) and PI3K/Akt/mTOR and MEK/Erk signaling pathways, regulating the expression of downstream effectors such as SOCS, Cyclin D2, BCL-2 and p27kip1 (reviewed in [109]). In T-ALL, PI3K activation is mandatory for IL-7-mediated p27kip1 down-regulation, Rb hyperphosphorylation, and BCL-2 upregulation, and is also required for cell size increase, expression of the glucose transporter Glut1, glucose uptake, and maintenance of mitochondrial integrity [110]. These findings indicate that PI3K is a major effector of IL-7–induced viability, metabolic activation, growth, and proliferation of T-ALL cells, highlighting the importance of developing targeting interventions directed to the IL-7R pathway. Because some B-ALL cases express functional IL-7Rs and respond to IL-7 as well [103][111], therapeutic targeting of IL-7R could be extended to B-cell leukemias. Several drugs capable of targeting IL-7R-activated routes are beginning to be explored clinically, with special emphasis on PI3K/Akt/mTOR, JAK/STAT5, and BCL-2 inhibition (reviewed in [32][112]).
Besides the accepted role of IL-7R in the maintenance and progression of T-ALL, several studies have pointed to a direct contribution of this signaling axis in the pathogenesis of T-ALL. Lymphoid malignancies develop spontaneously in both IL-7-transgenic and AKR/J mice that naturally overexpress IL-7Rα [113][114], and increased membrane IL-7R density leading to enhanced IL-7 signaling has been shown to cooperate with T-cell oncogenes in mouse T-ALL [115]. Conversely, progression of T-ALL patient-derived xenografts is hampered in IL-7-deficient mice [116]. More importantly, up to 10% of T-ALL cases harbor activating mutations in genes encoding different molecular effectors downstream of IL-7R signaling, including JAK1, JAK3 and STAT5B (reviewed in [71]), reinforcing the role of IL-7R in leukemia cell proliferation. Recently, direct proof has been provided that IL-7R signaling does, in fact, contribute to T-ALL pathogenesis [106]. Using loss-of-function approaches, it was formally established that IL-7R signaling is essential for Notch1 oncogenicity and T-cell leukemogenesis. The work also demonstrated that IL-7R expression contributes to human T-ALL LIC function, and revealed that IL-7R is a functional biomarker of T-ALL cells with LIC potential, a finding that was extended to B-ALLs expressing IL-7R [106]. The realization that IL-7R/IL-7 signaling directly contributes to T-ALL LIC activity has important clinical implications, as new therapeutic developments against leukemia mostly rely on molecular targets of LIC cells, which are finally responsible of disease relapse [51]. Therefore, targeting of normal IL-7R signaling represents a promising therapeutic approach for preventing T-ALL and, likely, B-ALL relapses, the major hurdle of ALL [117].
Overall, accumulated findings have placed IL-7R into focus as an oncogenic receptor for T-ALLs. This possibility was later confirmed by seminal studies showing that IL7R gain-of-function somatic mutations occur in around 10% of T-ALL and 1% of BCP-ALL patients at diagnosis [118][119]. These mutations are located in exon 5, and mostly in exon 6 encoding the juxtamembrane-transmembrane region of IL-7Rα. Exon 6 mutations consist of short insertions/deletions that introduce an unpaired cysteine residue capable of disulphide bond formation, allowing for pairing of two IL-7Rα chains and generation of an α-α homodimer, which triggers the activation of downstream IL-7R signaling cascades independently of IL-7. Decoding the signaling pathways selectively activated by oncogenic IL-7R, as compared to those induced by wild type IL-7R, will provide strong rationale for tailoring T-ALL (and B-ALL) therapies based on targetable IL-7R-related molecules that specifically eliminate leukemic cells, while preserving normal T-cell development and homeostasis. Yet, identification of such exclusive oncogenic molecules/pathways remains challenging. Thus, targeting of IL-7Rα itself has emerged as a promising alternative strategy for a great majority of T-ALL patients and some B-ALL patients, even for those harboring IL7R activating mutations.
Recent advances in clinical immunotherapy have now reached the IL-7R pathway. Several anti-IL-7Rα blocking mAbs are currently in Phase I or Phase IIa trials in patients with autoimmune diseases where IL-7R seems to play a critical role (reviewed in [120]), such as type 1 diabetes and multiple sclerosis, or Sjogren’s syndrome and inflammatory bowel disease, respectively (reviewed in [121]). In T-ALL, PDX models based on administration of anti-IL-7Rα mAbs and ADCs have provided promising results for therapeutic targeting of T-ALLs expressing either wild type or mutant IL-7Rs [122][123], and preclinical studies have also proved that mAb-mediated IL-7R targeting impairs B-ALL progression and metastasis [124]. Recently, Barata and co-workers [122] have generated a fully human mAb that recognizes human IL-7Rα and impairs IL-7/IL-7R-mediated signaling. The antibody sensitized T-ALL cells to treatment with dexamethasone, induced leukemia cell death in vitro through NK cell-mediated ADCC, and reduced leukemic burden in vivo. Conjugation with the antineoplastic toxin monomethyl auristatin E (MMAE) significantly increased the leukemia killing capacity of the mAb, validating the ADC approach. Additional work by Durum and coworkers showed that anti-IL-7Rα mAb-mediated ADCC had therapeutic efficacy against both relapsing and established disease in T-ALL preclinical models [123], leaving the door open for clinical trials evaluating the efficacy of this therapeutic strategy in unresponsive and relapsed patients.
An alternative, yet unavailable, strategy of targeting refractory T-ALLs expressing either wild type or mutant IL-7R may rely on the administration of anti-IL-7Rα CAR T cells. This approach is particularly challenging, considering the IL-7R-dependency of the lymphoid compartment and the finding that IL-7R is expressed in non-hematopoietic tissues as well [125][126]. This represents an important drawback, as potential therapeutic benefits may be counteracted by associated off-target toxicities and severe combined immunodeficiency [127]. Nevertheless, recent studies suggest that immunosuppression induced by treatment of mouse autoimmune arthritis with an anti-IL-7Rα ADC could be well tolerated clinically for a period of time [128], and very promising results have been reported from anti-IL-7R mAb clinical trials for autoimmune diseases [129]. Overall, current information on the major contribution of normal IL-7R signaling to the onset, maintenance, and progression of acute leukemias opens a new and major area of research to develop and validate novel therapies focusing on the IL-7R pathway.
2.3. CD44
CD44 is a type I single-span transmembrane glycoprotein that functions as a receptor for the glycosaminoglycan hyaluronan (hyaluronic acid, HA) [130][131], a major component of the extracellular matrix (ECM). The CD44 gene contains 19 exons in humans, and generates a variety of tissue-specific isoforms different in size through alternative splicing and N—and O-glycosylation [132][133][134]. In the hematopoietic system, the CD44 standard isoform (CD44s), containing 10 exons, is the most abundant, whereas variant isoforms (CD44v), which contain different exons assembled in the variable domain at the juxtamembrane region of the receptor, present a more restricted expression [135]. CD44v isoforms are expressed in discrete populations of epithelial cells and in some hematopoietic cell subsets, particularly during development; in several types of carcinoma [136], and after lymphocyte activation [137]. Many studies have focused on the CD44v6 isoform because it is frequently upregulated in cancer cells of epithelial origin, where it plays a role in migration, metastasis and chemoresistance (reviewed in [138]) and has been associated with poor prognosis [139][140].
The first described function for CD44 was as a molecule implicated in lymphocyte homing into lymph nodes. Afterward, CD44 has been shown to play a prominent role in the anchoring of both hematopoietic precursors [141][142] and leukemic cells [143] within the bone marrow niche. Notably, CD44 is also expressed during intrathymic T-cell development, both in human and mouse, where it defines different progenitor stages with singular intrinsic linage potentialities. CD44 is expressed at high levels in the earliest human CD34+ T-cell precursors seeding the thymus, and its expression is downregulated upon T-cell commitment and during T-cell development, while it is maintained in myeloid-primed intrathymic progenitors with dendritic cell potential [144][145][146].
Importantly, CD44 is one of the most studied markers of cancer-initiating cell (CIC), with a key function in the maintenance of CIC-associated properties in several solid tumors (reviewed in ). However, its contribution to LIC activity in hematologic cancers, although suggested, has not been formally proven until very recently. By using a novel mouse model of human T-ALL pathogenesis [147], this work showed that CD44 is a direct transcriptional target of NOTCH1, which is upregulated in NOTCH1-induced preleukemic blasts, facilitating their engraftment into the bone marrow niche, and supporting the LIC potential of T-ALL. These results indicate that CD44 upregulation is a key event of T-cell leukemogenesis, a finding that concurs with the fact that T-ALL patients commonly display an aberrant expression of CD44 in leukemic blasts [148]. Although no link of CD44 expression with prognosis and overall survival of T-ALL patients has been established, CD44 contributes to T-ALL maintenance and progression, likely by regulating LIC potential [149], as proved by anti-CD44 mAb administration in a PDX T-ALL model. CD44 expression is also upregulated in Notch1-induced T-ALL leukemic cells treated with chemotherapeutic drugs, such as doxorubicin and dexamethasone, and contributes to T-ALL chemoresistance by modulating intracellular drug efflux [150]. Consequently, CD44 may represent a valuable target for new therapies against relapsed or chemoresistant T-ALLs.
Promising results have been obtained in r/r AML patients treated with an anti-CD44 blocking antibo[151]. In PDX preclinical settings, grafting of AML patient cells into mouse bone marrow can be blocked by administration of an activating anti-CD44 mAb [152], which targets AML leukemic stem cells by inducing their differentiation [153] or by increasing apoptosis [154][155], leading to AML eradication. An ADC consisting of an anti-CD44 antibody (Bivatuzumab) bound to mertansine was tested in phase I clinical trials for head and neck and esophagus squamous cell carcinomas [156][157] and for breast cancer [158], but the trials had to be terminated before progression to phase II due to severe skin toxicities. Recent trials have also tested a commercialized anti-CD44 mAb developed by Roche (RO5429083) for metastatic and/or advanced solid tumors and AML but, although completed, the trial results have not been published yet. Besides mAbs, recent development of anti-CD44 CARs has opened new opportunities for treatment of CD44-expressing cancers. In particular, anti-CD44v6 CAR T-cell immunotherapy trials for multiple cancers (NCT04427449), r/r AML, MM (NCT04097301) and breast cancer (NCT02046928) are ongoing, representing promising strategies worth to be extended to T-ALL and other CD44+ T-cell malignancies, once safety concerns are solved and efficacy proved.
CD30 belongs to the tumor necrosis factor receptor (TNFR) superfamily [159] and, although initially described as an antigen expressed on Hodgkin´s lymphoma (HL) cells, it was later found to be expressed in other hematological malignancies such as anaplastic large cell lymphomas (ALCL), CTCL, PTCL, adult T-cell leukemia/lymphoma, and diffuse large B cell lymphomas (DLBCL) [160][161]. In normal cells, CD30 is expressed by T helper (Th) cells [162] (preferentially Th2-type cytokine secreting cells [163]), by a subset of CD8+ peripheral blood lymphocytes [164], and by the B-1 subset of B lymphocytes [165]. Upregulation of CD30 expression was documented after stimulation or viral infections [i.e., Epstein-Barr virus (EBV), human T-cell leukemia virus (HTLV) and human immunodeficiency virus (HIV)] in lymphoid cells. Although CD30 induction was described after activation of both HSPCs [166] and T cells [167], the expression levels observed were lower than those displayed by tumor cells. Consequently, anti-CD30 treatment has proven to be effective in eliminating tumor cells without affecting normal lymphopoiesis in preclinical assays, which pointed to CD30 as a very attractive candidate for immunotherapeutic targeting. Anti-CD30 immunotherapy has been successfully applied as ADC (brentuximab vedotin) to r/r adult T-cell leukemia/lymphoma patients [168], and r/r CD30+ lymphoma patients [169], showing an especially good response in Hodgkin lymphoma and systemic ALCL cases. In addition, anti-CD30 directed CAR T cells have been tested in clinical trials for r/r HL or ALCL, reporting acceptable tolerability but modest effectiveness [170][171]. To date, several trials are open to assess safety and efficacy of CD30 CAR T cells in different hematological malignancies, although none of them include T-ALL patients. However, CD30 expression (>20% positive cells as assessed by flow cytometry) has been detected in 13/34 cases of T-ALL and in 6/44 cases of B-ALL [172]. Furthermore, CD30 expression is increased in bone marrow samples of T-ALL patients treated with high-dose chemotherapy, indicating that application of anti-CD30 immunotherapy should be considered when designing new trials for CD30+ r/r T-ALL patients.
CD99 (MIC2) is an O-glycosylated transmembrane protein expressed on leukocytes and activated endothelium that mediates cell adhesion [173], T-cell activation [174], and transendothelial migration of different cell types, including leukocytes [175]. The CD99 gene encodes two different isoforms with differential expression patterns and functions. Anti-CD99 monoclonal antibodies were able to induce caspase-independent cell death of AML cell lines and primary blasts [176], T-cell lines [177] and BCP-ALL cells bearing the TEL/AML1 fusion [178]. T-ALL cells have been shown to express higher levels of CD99 than hematopoietic stem cells and normal T cells [179][180], and detection of CD99 expression by flow cytometry was demonstrated useful for revealing MRD in T-ALL patients [179][181], pointing to CD99 as a promising target for immunotherapy. However, LIC frequency assays indicated that CD99+ T-ALL fraction was not enriched in cells with LIC potential [179], thus precluding the suitability of CD99 as therapeutic target for relapsed T-ALL. A potential new therapeutic option based on CD99 targeting has emerged from in vitro studies showing that treatment of T- and B-cell lines with anti-CD99 antibodies leads to upregulation of heat shock protein 70 (HSP70) (a prerequisite for NK-dependent lytic activity), making leukemic cells susceptible to NK-cell cytotoxicity [182]. Thus, reassessment of CD99 as a candidate for alternative therapeutic strategies against T-ALL seems realistic.
Recent advances in targeted immunotherapies for B-cell malignancies have engendered unprecedented expectations for the successful treatment of T-ALL patients, with the challenge still pending on establishing protocols for clinical management of associated side effects. Nevertheless, current progress, particularly on 1) the application of universal off-the-shelf CAR T cells that prevent fratricide; 2) the incorporation of suicide genes allowing for CAR T-cell elimination after tumor eradication and T-cell immunodeficiency reversion; and 3) the discovery of increasingly specific molecular targets proved critical for disease progression in preclinical models, has tilted the balance between risks and benefits towards the use of immunotherapy for r/r T-ALL. Still, avoidance of associated adverse effects demands further efforts for the identification of new unique T-ALL antigens absent on normal T cells that guarantee the safe and effective application of these strategies.





