1000/1000
Hot
Most Recent
 +1 point
+1 point
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system and its pathophysiology is characterized by a progressive blood-brain barrier dysfunction accompanied by infiltration in the central nervous system of peripheral pathogenic immune cells and inflammatory mediators leading to demyelination, axonal damage, and dysfunction and/or loss of synapses. Accumulating evidence highlights blood and cerebrospinal fluid (CSF) derived extracellular vesicles (EVs) as potential biomarkers of MS disease stages and of response to treatment. In particular, EVs released from blood–brain barrier (BBB) endothelial cells, platelets, leukocytes, myeloid cells, astrocytes, and oligodendrocytes seem to be involved in the pathogenesis of MS and of its rodent model experimental autoimmune encephalomyelitis. Further research is necessary to validate these observations and the screening of specific EVs subsets based on their cargo and membrane compositions associated to specific MS pathophysiological mechanisms might help guiding MS diagnosis, prognosis, and response to therapy.
MS is a chronic inflammatory disease of the central nervous system (CNS), characterized by a wide variety of neurological symptoms including muscle weakness, sensory, visual and cerebellar deficits, cognitive impairments, and psychic symptoms such as fatigue and depression [2]. The clinical course of MS is classified by McDonald’s diagnostic criteria in two different phenotypes: relapsing–remitting and progressive [2]. The so-called relapsing remitting MS (RRMS) is the most common phenotype, and it is characterized by acute episodes of neurological deficits (relapse) followed by a return to baseline function (remission) of clinical symptoms. Over a long term follow-up, 15–30% of RRMS patients develops progressive disability, and this phenotype is classified as secondary progressive multiple sclerosis (SPMS). About 15% of MS patients directly develops a progressive phenotype from the outset and are classified as primary progressive multiple sclerosis (PPMS) patients [3]. Clinical classification also provides clinically isolated syndrome (CIS), a single clinical event compatible with MS that could both evolve in a RRMS or remain isolated, and radiologically isolated syndrome (RIS), an incidental finding of radiological signs of disease in the absence of clear clinical activity [4].
The etiology of MS is still unknown, but it is evident that a complex interaction between environmental, genetic, and epigenetic factors triggers an autoimmune reaction against the CNS compartment [2][1]. The most accredited hypothesis is that peripheral T and B lymphocytes, primed against a still-unknown antigen, drive a cross-reaction against CNS epitopes including oligodendrocytes proteins, such as myelin basic protein, proteolipid protein, and myelin oligodendrocyte glycoprotein (MOG). Animal modeling, despite several limitations, has been crucial to understand MS pathogenesis. In particular, the experimental autoimmune encephalomyelitis (EAE) has greatly contributed to our understanding of autoimmunity and of inflammation-induced neurodegenerative processes [5]. A hallmark of MS pathophysiology is a progressive BBB dysfunction that causes infiltration in the CNS of peripheral pathogenic T and B cells, antibodies, monocytes, and inflammatory mediators. The chain of inflammatory reaction triggered by infiltrating leucocytes leads to demyelination, axonal damage, and synaptic loss and dysfunction, named synaptopathy, ultimately resulting in a prominent neurodegeneration [1][6][7]. Moreover, pro-inflammatory mediators, including interferon-γ (INF-γ), interleukin-1β (IL-1β), and particularly, tumor necrosis factor-α (TNF-α), released by peripheral leucocytes and activated microglia and astrocytes, favorite adhesion of activated leucocytes on the endothelium by overexpression of nitric oxide and adhesion molecules (VCAM-1, E-selectin, and CD31/PECAM-1) [8]. Such inflammatory milieu leads to endothelial release of metalloprotease and proteolytic enzymes that contribute to BBB disruption, further increasing the trafficking of autoreactive T and B cells, antibodies, monocytes, and inflammatory mediators from vessels into the CNS. Radiological evidence of a leaky BBB is provided by the contrast (gadolinium; gad+) enhancement of active plaques in magnetic resonance imaging (MRI) [9]. White matter neuroinflammatory foci are indeed easily detectable during the acquisition of MRI, appearing as hyper-intense areas called “plaques” or demyelinating lesions. The demyelination process is partially counteracted by proliferation and migration of oligodendroglial precursor cells (OPCs, NG2+) to the lesion site, where they differentiate in mature oligodendrocytes (OLGs) and form the new myelin sheath [7]. However, in MS patients OPCs are often detained at the plaque edge, or they may differentiate into malfunctioning premyelinating OLGs [10]. The interplay between OPCs and glial cells seems to cover a crucial role in the remyelination process. In particular, a pro-regenerative microenvironment can be produced by a complex interaction between mesenchymal stem cells (MSC), microglia, astrocytes, and IL-4-releasing macrophages [11][12]. The presence of MSCs during neuroinflammation influences the release of IL-4, a cytokine involved into remyelinating processes, and favorites the expression of pro-regenerative genes by activated microglia [13]. On the other hand, in the absence of MSCs, primary microglia express elevated levels of pro-inflammatory genes and mediators, such as IL-1α, C1q, IL-1β, inducible nitric oxide synthase (iNOS) [13]. The establishment of a chronic inflammatory state, caused by a microenvironment enriched with activated microglia releasing pro-inflammatory cytokines, such as TNF-α [13], gradually impairs remyelinating processes as a result of altered OPC activation and recruitment to demyelinating lesions [12]. This process leads to a consistent axonal loss and is associated to clinical impairment and the progression of disease.
The MS brain is also affected by another degenerative but potentially reversible phenomenon, namely inflammatory synaptopathy. Clinical research using transcranial magnetic stimulation (TMS) [14], pre-clinical studies conducted on an EAE model [15], and more recently chimeric ex-vivo models [6][16] have highlighted the presence of an inflammation-dependent decrease in the gamma-aminobutyric acid (GABA) ergic tone and an increase in the glutamatergic transmission in several MS/EAE brain areas. Such a synaptic transmission unbalances results in a diffuse synaptic dysfunction and loss that is mediated by pro-inflammatory molecules released by peripheral immune system cells, microglia, and astroglia [6][14]. On the other hand, synaptic plasticity events may intervene as a compensatory mechanism to overcome synaptic damage, becoming exhausted in the MS progressive forms. The consequence of a long-lasting synaptic imbalance is an excitotoxic damage that triggers neurodegenerative processes [6][14].
Accumulating evidence highlights serum/plasmatic and CSF EVs as potential biomarkers of MS disease stages and of response to treatment (Table 1).
Table 1. Extracellular vesicle (EV) classification and their potential role in multiple sclerosis (MS).
| EVs Cellular Origin | Surface Marker | Functional Implication | Detection Levels | References | Study Size |
|---|---|---|---|---|---|
| Serum/Plasma | |||||
| Endothelial cells | CD31+ | Acute BBB disruption and contribution in Gad+ MRI active lesions |
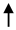 E-MS RRMS E-MS RRMS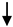 R-MS; SPMS; HC R-MS; SPMS; HC |
Minagar et al., 2001 [17] | 48 HC; 30 E-RRMS; 20 R-RRMS |
| Jy et al., 2004 [18] | 35 HC; 30 E-RRMS; 20 R-RRMS | ||||
| Alexander et al., 2015 [19] | 36 HC; 44 RRMS; 16 SPMS |
||||
| CD4+ and CD8+ T-lymphocytes activation | Wheway et al., 2014 [20] | ||||
| CD51+ | Chronic endothelial injury | E-MS; R-MS HC |
Minagar et al., 2001 [17] | 48 HC; 30 E-RRMS; 20 R-RRMS | |
| CD54+ CD62E CD106+ |
Monocytes conjugates for endothelial adhesion | E-MS R-MS; HC |
Jy et al., 2004 [18] | 35 HC; 30 E-RRMS; 20 R-RRMS | |
| Jimenez et al., 2005 [21] | 10 HC; 11 E-RRMS; 9 R-RRMS |
||||
| Monocytes | CD14+ | Acute endothelial injury | R-RRMS HC and SPMS |
Saenz-Cuesta et al., 2014 [22] | 20 HC; 13 SPMS 64 R-RRMS |
| Leukocytes | CD45+ | Acute endothelial injury | R-RRMS HC and SPMS |
Saenz-Cuesta et al., 2014 [22] | 20 HC; 13 SPMS 64 R-RRMS |
| Platelets | CD62p CD41+/ CD61+ |
Platelets activation and leukocytes interaction with damaged endothelium | R-MS (RRMS) HC and SPMS |
Saenz-Cuesta et al., 2014 [22] | 20 HC; 13 SPMS 64 R-RRMS |
| Sheremata et al., 2008 [23] | 92 HC; 33 R-RRMS | ||||
| CD42b+ | Incremented experimental BBB permeability (except for CIS) |
MS (PMS; RMS; CIS) HC |
Marcos-Ramiro et al., 2014 [24] | 49 HC; 23 SPMS; 51 RRMS; 12 CIS; 9 PPMS |
|
| Cerebrospinal Fluid | |||||
| Microglia/Macrophage | IB-4 | Acute BBB disruption and contribution in Gad+ MRI active lesions |
E-MS (CIS, RRMS) R-MS (RRMS); HC |
Verderio et al., 2012 [25] | 13 HC; 39 R- RRMS 28 E-RRMS; 28 CIS |
| T-cells | CCR3/ CCR5 CD4/ CCR3 CD4/ CCR5 |
Acute BBB disruption and contribution in Gad+ MRI active lesions |
E-MS (RRMS) R-MS (RRMS) |
Geraci et al., 2018 [26] | 10 R-RRMS; 13 E-RRMS |
Abbreviations: BBB (blood–brain barrier); E-MS (exacerbated-MS); R-MS (remission-MS); HC (healthy controls); RRMS (relapsing–remitting MS); SPMS (secondary progressive MS); CIS (clinically isolated syndrome); OPC (oligodendroglial precursor cells). Up and down arrows refer to high and low levels of EVs, respectively.
In the attempt to identify blood-derived EVs as disease stage biomarkers, important associations emerged between EVs, mostly derived from platelet-(CD61+), leukocyte- (CD45+), monocyte (CD14+) [27], and endothelium-(CD51+ and CD31/PECAM-1+) cells [17] and disease activity, progression, and drug response. One of the first studies, conducted by Minagar in 2001, showed that plasma levels of CD31/PECAM-1+ EVs released by endothelial cells were significantly higher in RRMS patients during the active phase of disease compared to non-active RRMS patients and healthy control (HC) subjects. A positive correlation with active (gad+) lesions in MRI [17] emerged, suggesting that high levels of EVs detectable in the patients’ plasma may anticipate a radiological relapse of disease [17]. These results are in line with the observation that the CD31/PECAM-1 adhesion molecule is strictly involved in transendothelial migration of leukocytes during the inflammatory processes. Conversely, endothelial EVs CD51+ (integrin alpha-V) remained elevated during both exacerbation and remission, thus appearing to be a marker of chronic damage more than endothelial disruption [17]. Other clinical studies showed that endothelial EVs were significantly increased in the serum of both SPMS and RRMS patients, with higher levels in relapsing–remitting than progressive forms, thus reflecting a status of active peripheral inflammation rather than chronic neurodegeneration [19][22][28]. Interestingly, elevated plasma levels of conjugates between endothelial EVs and monocytes directly correlated with MRI gad+ lesions in a court of RRMS patients [18]. In vitro studies suggested that the binding of endothelial EVs to monocytes might promote their activation by enhancing monocytes’ migration through an endothelial monolayer [18]. The association between active MRI lesions and plasma levels of EVs was further supported by Jimenez and colleagues that observed an increase in endothelial EVs in MS patients during the clinical relapse phase compared to remission [21], suggesting endothelial EVs as potential biomarkers of BBB damage. Besides EVs derived from endothelial cells, Sheremata and colleagues, pointed out to an aberrant activation of platelets in MS as a secondary effect of a chronic endothelial damage showing a high count of CD41+ platelet EVs in MS plasma compared to HC [23]. These data were supported by the observation that RRMS in remission present higher levels of platelet EVs plasma levels, together with monocyte- and leukocyte-derived EVs, in comparison to SPMS and HC [22]
Previous studies in MS have assessed the content of circulating EVs and have demonstrated significant alterations in miRNA profiles and relationships to disease course [29][30][31] (Table 2). Ebrahimkhani and colleagues demonstrated that serum—exosome cargo of microRNA, with no specification of its cellular origin—was different in RRMS (miR-15b-5p, miR-451a, miR-30b-5p, miR-342-3p) and progressive MS patient sera (miR-127-3p, miR-370-3p, miR-409-3p, miR-432-5p) in comparison to HC [29]. Selmaj and colleagues observed a significant reduction in several serum-exosomal miRNAs (hsa-miR-122-5p, hsa-miR-196b-5p, hsa-miR-301a-3p, and hsa-miR-532-5p) during relapse in RRMS [31]. These miRNAs were also decreased in patients with a gadolinium enhancement in brain MRI. The authors also assessed in vitro the secretion of these miRNAs by peripheral blood mononuclear cells (PBMC) and observed a significant impairment in RRMS. Specific miRNA have also been implicated in potentially pathogenic effects on the immune system in MS [32][33] and therapeutic response [34].
Table 2. Classification of miRNA content in EVs involved in MS.
| miRNA | Detection Level | Study Size | Functional Implication | Reference |
|---|---|---|---|---|
| miR-15b-5p | In RRMS vs. HC |
14 RRMS 11 S/PPMS 11 HC |
Targets FGF-2 implicated in demyelination and remyelination | Ebrahimkhani et al., 2017 [29] |
| miR-451a | In RRMS vs. HC |
14 RRMS 11 S/PPMS 11 HC |
Regulator of oxidative stress | Ebrahimkhani et al., 2017 [29] |
| miR-30b-5p | In RRMS vs. HC |
14 RRMS 11 S/PPMS 11 HC |
Neuro-axonal injury | Ebrahimkhani et al., 2017 [29] |
| miR-342-3p | In RRMS vs. HC |
14 RRMS 11 S/PPMS 11 HC |
Neuro-axonal injury | Ebrahimkhani et al., 2017 [29] |
| miR-127-3p | In S/PPMS vs. HC |
14 RRMS 11 S/PPMS 11 HC |
Ebrahimkhani et al., 2017 [29] | |
| miR-370-3p | In S/PPMS vs. HC |
14 RRMS 11 S/PPMS 11 HC |
Ebrahimkhani et al., 2017 [29] | |
| miR-409-3p | In S/PPMS vs. HC |
14 RRMS 11 S/PPMS 11 HC |
Ebrahimkhani et al., 2017 [29] | |
| miR-432-5p | In S/PPMS vs. HC |
14 RRMS 11 S/PPMS 11 HC |
Ebrahimkhani et al., 2017 [29] | |
| miR-122-5p | In remission RRMS vs. HC; in relapse RRMS vs. remission RRMS |
30 Remission- RRMS 33 Relapse- RRMS 32 HC |
Targets STAT3 and AHR (not validated), regulators of differentiation of Th17 and immunosuppressive T cells | Selmaj et al., 2017 [31] |
| miR-196b-5p | In relapse RRMS vs. HC; in relapse RRMS vs. remission RRMS |
30 Remission- RRMS 33 Relapse- RRMS 32 HC |
Targets STAT3 and AHR (not validated), regulators of differentiation of Th17 and immunosuppressive T cells | Selmaj et al., 2017 [31] |
| miR-301a-3p | In relapse RRMS vs. HC |
30 Remission- RRMS 33 Relapse- RRMS 32 HC |
Targets STAT3 and AHR (not validated), regulators of differentiation of Th17 and immunosuppressive T cells | Selmaj et al., 2017 [31] |
| miR-532-5p | In relapse RRMS vs. HC; in relapse RRMS vs. remission RRMS |
30 Remission- RRMS 33 Relapse- RRMS 32 HC |
Targets STAT3 and AHR (not validated), regulators of differentiation of Th17 and immunosuppressive T cells | Selmaj et al., 2017 [31] |
| Let-7i | In MS vs. HC |
4 MS 4 HC |
Inhibition of Treg cells differentiation from naive CD4+ T cells | Kimura et al., 2018 [32] |
| miR-146a-5p | Detected in CSF exosomes | MS = 10 | Synaptic alterations in in vitro experiments | Prada et al., 2018 [35] |
| miR-219a-5p | Artificially enriched exosomes | EAE mice | Maturation of OPCs; clinical score improvement | Osorio-Querejeta et al., 2020 [36] |
MiRNA detected in EVs derived from serum and plasma of MS patients. Abbreviations: HC (healthy controls); RRMS (relapsing–remitting MS); SPMS (secondary progressive MS); PPMS (primary progressive MS); FGF-2 (fibroblast growth factor-2); STAT3 (signal transducer and activator of transcription 3); AHR (aryl hydrocarbon receptor); EAE (experimental autoimmune encephalomyelitis); OPCs (oligodendrocyte precursor cells). Up and down arrows refer to high and low levels of miRNA, respectively.
One previous study evaluated the protein cargo of circulating EVs and noted alterations in myelin associated proteins [31]. Plasmatic and CSF PBMC-derived exosomal content of MOG directly correlated with radiological relapse of disease in RRMS and SPMS patients. The authors suggested that this peripheral release of EVs containing myelin antigen may sustain inflammation, peripheral activation, and migration of immune cells against CNS oligodendrocytes perpetuating the anti-myelin immune reactions [31].
Due to its proximity to the CNS, the most direct source of biomarkers is CSF [37][38]. Biochemical and molecular analysis of the CSF has been indeed compared to a liquid biopsy of CNS, but lumbar puncture to collect this precious biological fluid is an invasive procedure not applicable for the follow-up of MS patients [39]. Furthermore, the relative low amount of EVs as well as the small volume of CSF available have made EVs isolation from CSF very challenging. However, researchers are developing new techniques to make CSF analysis a routine part of optimal MS clinical management and to find alternative and less invasive approaches [40]. In this regard, a recent work has compared the levels of microvesicles in CSF and in tears, supporting the latter as intriguing possible samples for the study of EVs [41].
Among the data obtained in the CSF, early observations by Scolding and colleagues revealed the presence of vesicles in the CSF of MS patients [42]. More recently, Verderio et al. confirmed these data by showing an overproduction of EVs in CSF in subjects with a diagnosis of CIS or MS versus HC [25]. MVs display neuronal, astrocytic, oligodendroglial, or microglia/macrophage markers, thus indicating that they originate from all these brain cells. By focusing on MVs of myeloid origin the authors found a positive correlation between macrophage/microglia EVs levels and radiological activity of MS disease. Notably, they showed a high degree of sensitivity and specificity in the distinction between CIS and HC and between active and non-active patients [25], suggesting macrophagic/microglial EVs as optimal biomarkers of neuroinflammation.
An important change in the number of CSF EVs and in their surface marker expression during active phases of MS was confirmed in a recent study, in particular a CSF EV’s increase was detected in patients affected by MS during clinical relapse; this finding was associated with a decrease in the number of CD19+/CD200+ (naïve B cells) EVs. Furthermore, an association emerged between gadolinium-enhanced MRI lesions in the CNS and the increase in the number of CCR3/CCR5 (subset of CD8 memory T cells), CD4/CCR3 (Th2 cells), and CD4/CCR5 (Th1 cells) CSF EVs, emphasizing again EVs as a pivotal and promising biomarker of inflammatory activity [26].
In another study, Masvekar and colleague did not find a difference between levels of EVs deriving from cell apoptosis (apoptosomes) isolated in CSF of RRMS (active and non-active) patients and HC, suggesting that apoptotic bodies are not appropriate as disease biomarkers [43], but further validations are necessary.
EVs have been proposed as an accessible predictive marker of the pharmacological response to disease-modifying therapies (DMT) in MS. Several studies contributed in this sense to expand current knowledge. Preclinical and clinical studies based on treatment with the second line oral drug fingolimod (FGM), the founder of sphingosine 1-phosphate receptor modulator drugs, showed a considerable reduction in microglia-derived EVs in the CSF of both active and non-active RRMS and PPMS patients and in EAE mice [25]. Such an effect was expected considering that FGM is a specific inhibitor of sphingomyelinase acid potentially involved in EV release. In the long term, a changing of the EVs content was also observed, consisting in a differential expression of miRNA and other compounds implicated in recovery from damage. In another study conducted on a court of active and non-active RRMS patients under FGM treatment, Zinger and colleagues observed that endothelial EVs (CD 105+) were significantly higher in MS patients relatively to HC and that FGM treatment was able to reduce their levels, reaching values similar to HC. In support of these results, the authors showed a reduced formation of surface blebs in human brain endothelial cells pre-cultured for 24 h with FGM and exposed to TNF-α for 18 h [44]. Curiously, the author observed that EVs derived from B cells (CD19+) were less in MS untreated patients compared to HC and that were significantly increased after FGM administration [44]. This differential effect of FGM on EVs release was not investigated. Similarly, in a study conducted by Sáenz-Cuesta and colleagues, FGM treatment induced an early enhancement of the EVs content in the plasma, mainly represented by platelet-derived (CD61+), leukocyte-derived (CD45+), and monocyte-derived (CD14+) EVs. The author also showed that the inhibitory role on lymphocyte activation exerted by circulating EVs was reduced following FGM treatment and observed a modulation of the EV miRNA cargo (miRNA not specified) [27]. Of note, such effects were observed after 5 h from the treatment and were interpreted by the authors as a consequence of the microenvironmental changing induced by FGM; lymphocyte arrest in lymph nodes induced by FGM may result in a low inflammatory status at peripheral level, where more EVs with a low regulatory profile are released. Based on all this evidence, FGM treatment certainly exerts different effects on EV depending on the cell-type origin, MS status, and duration of the treatment. Multiple mechanisms and concomitant factors might contribute to providing different effects on EVs, making difficult to compare results among these diverse studies. Furthermore, the lack of a deep knowledge of the EV function and of standardization in the methodology does no help in clarifying this complex scenario.
Similarly, it has been observed that IFN-β, a first-line treatment for RRMS patients, was able to stabilize the injured endothelium in association with a decrease in CD31+/PECAM-1+, CD54+, and CD146/intercellular adhesion molecule (ICAM-1) + EVs [40]. Conversely, in a different work, RRMS patients treated with IFN-β or natalizumab had significant higher counts of three EV subtypes (platelets-, total leukocytes-, or monocytes-derived) compared with untreated patients [22]. The mechanisms underlying such effects were not fully elucidated. Finally, a recent study conducted on a population of non-active RRMS patients treated with IFN-β or drug naïve showed a different exosomal miRNA profiling between RRMS treated and untreated patients, suggesting exosomal miRNA cargo as a possible prognostic tool of drug response [34].
Altogether, these data clearly highlight the relevance of EVs as potential biomarkers for diagnosis and therapeutic response in MS, however validated and reproducible results are still missing. Future research toward the screening of specific EVs subsets based on their cargo and membrane compositions associated to specific MS pathogenetic mechanisms might help guiding MS diagnosis, prognosis, and response to therapy.





