1000/1000
Hot
Most Recent
 +1 point
+1 point
Peritoneal dialysis (PD) is a current replacement therapy option for end-stage renal disease (ESRD) patients until renal transplantation can be achieved. One important problem in long-term PD patients is peritoneal membrane failure. The mechanisms involved in peritoneal damage include activation of the inflammatory and immune responses, associated with submesothelial immune infiltrates, angiogenesis, loss of the mesothelial layer, and collagen accumulation in the submesothelial compact zone. These processes lead to fibrosis and loss of peritoneal membrane function. Among the inflammatory mediators involved in peritoneal damage, cytokine IL-17A has recently been proposed as a potential therapeutic target for chronic inflammatory diseases, including chronic kidney diseases (CKD). Experimental studies demonstrated that IL-17A blockade ameliorated peritoneal damage caused by exposure to PD fluids. This article provides a comprehensive review of recent advances in the role of IL-17A in peritoneal membrane injury during PD and other PD-associated complications.
Few studies have explored the effect of Interleukin-17A (IL-17A) in vivo in the peritoneum. In mice, a single intraperitoneal administration of IL-17A resulted in a rapid increase of local levels of Granulocyte-colony stimulating factor (G-CSF) and selective neutrophil accumulation [1]. In another study, intraperitoneal IL-17A induced submesothelial inflammation, together with the presence of monocytes, CD3+ and CD4+ T lymphocytes, and neutrophils, observed at 10 days. Moreover, weekly IL-17A intraperitoneal injections for 35 days induced peritoneal fibrosis characterized by peritoneal membrane (PM) thickness associated with fibronectin deposition and expression of myofibroblast markers, such as fibroblast-specific protein 1 (FSP-1) and α-smooth muscle actin (α-SMA) [2]. These data suggest a direct deleterious effect of long exposure to IL-17A in the peritoneum, contributing to the peritoneal damage induced by PD (Figure 1).
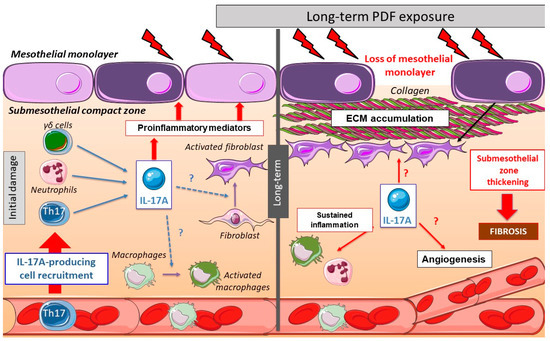
Figure 1. Peritoneal changes due to long-term peritoneal dialysis fluids (PDF) exposure: initially, chronic PDF exposure causes the recruitment of inflammatory cells into the submesothelial zone. Among the infiltrating immune cells, there are several IL-17A-secreting cells, such as T helper (Th)17 cells, γδ T cells, neutrophils, and others. The local production of IL-17A triggers the release of additional pro-inflammatory mediators by infiltrating cells and resident peritoneal cells, including cytokines and chemokines, therefore contributing to the amplification of the inflammatory response. In long-term PDF exposure, the loss of mesothelial monolayer and submesothelial thickness is associated with elevated peritoneal IL-17A levels. This cytokine could also potentially promote fibrosis and angiogenesis in the peritoneum.
Local tissue damage triggers an inflammatory response characterized by chemokine secretion and immune cell recruitment. Mediators secreted from immune cells eventually drive tissue regeneration and a transient local profibrotic response. However, failure of the reparative process may lead to persistent inflammation, excessive extracellular matrix (ECM) deposition, and fibrosis [3]. There is in vivo evidence supporting a profibrogenic role of IL-17A in pathological conditions associated with inflammation [4]. Thus, in cultured dermal vascular smooth muscle cells and fibroblasts from systemic sclerosis patients, IL-17A stimulated proinflammatory responses, ECM protein secretion, proliferation, and migration [5][6], supporting the profibrotic role of IL-17A.
Accordingly, the repeated exposure of the peritoneum to PDF elicits several cellular and molecular responses in the PM, including activation of an inflammatory response, production of cytokines and chemokines, and recruitment of inflammatory cells (Figure 1). As commented above, preclinical data suggest that long-term exposure to PDF induced the presence of submesothelial IL-17A-secreting cells. Moreover, IL-17A could activate peritoneal cells to upregulate some proinflammatory cytokines, like IL-6 or monocyte chemoattractant protein-1 (MCP-1), which contribute to persistent inflammation. This inflammatory response could also trigger the production of profibrotic factors, indirectly contributing to fibrosis (Figure 1).
IL-17A binding to its receptor in mesothelial cells can induce pro-inflammatory responses. In cultured human mesothelial cells, IL-17A activated the canonical NF-κB pathway and downstream cytokines, including G-CSF [1] and the C-X-C chemokine GROalpha (also known as CXCL1) [7]. This pro-inflammatory response was increased in the presence of TNF-α [1]. In high-glucose conditions, as occurs in response to conventional PDF exposure, mesothelial cells increased the production of proinflammatory and profibrotic factors. Interestingly, high glucose activated the TLR4/MyD88/NF-κB signaling pathway to induce inflammatory mediators, as MCP-1, in mesothelial cells [8]. However, the role of TLR4 in IL-17A responses has not been evaluated.
The exposure of peritoneal mesothelial cells to PDF results in Mesothelial-to-Mesenchymal Transition (MMT), a process characterized by phenotype alterations that induce a transition from an epithelial to a mesenchymal migrative phenotype [9][10][11][12][13]. The mesothelial cells begin to lose basolateral polarization and the expression of epithelial markers like cytokeratins, E-cadherins, and cell-junction proteins and acquire mesenchymal markers such as α-SMA, vimentin, vascular endothelial growth factor (VEGF)-A, Snail, collagens, and fibronectin [10][14][15][16][17][18][19]. Different mediators are involved in PDF-induced MMT, including advanced glycation end products (AGEs) [15], endotelin-1, growth factors, such as transforming growth factor-beta 1 (TGF-β1), VEGF, Gremlin-1 (GREM1), and connective tissue growth factor (CTGF/CCN2) as well as pro-inflammatory cytokines like IL-6 [10][11][16][17][18][19]. Although to date there is no definitive reported data, it is tempting to speculate that IL-17A may be a direct triggering stimulus of the MMT process (Figure 2).
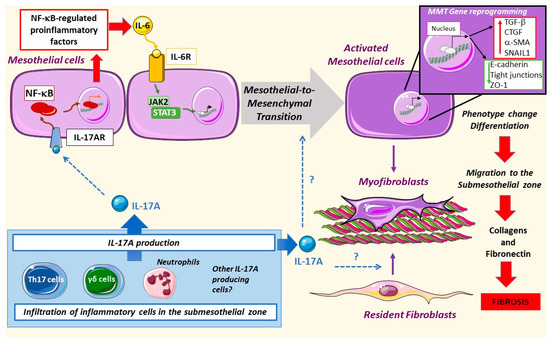
Figure 2. Mesothelial-to-mesenchymal transition in peritoneal damage by PDF: IL-17A produced by different cells can activate the nuclear factor-κB (NF-κB) pathway in mesothelial cells, driving the expression of regulated factors, such as IL-6. This cytokine can activate the Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway leading to mesothelial-to-mesenchymal transition (MMT). Moreover, mesothelial cells change their pool gene expression as well as phenotype, increasing the motility of these cells and the deposition of collagens and fibronectin, thus promoting fibrosis. IL-17A might also activate resident fibroblasts as well as trigger MMT directly, but these processes have not been yet explored.
Different signaling systems are involved in MMT. IL-17A promotes the peritoneal expression of IL-6 [2], which in a paracrine or autocrine manner binds and activates its receptor in mesothelial cells, engaging the JAK2/STAT3 pathway [20] and triggering MMT. On the other hand, TGF-β1 activation of the Smad pathway promotes the expression of profibrogenic proteins and myofibroblast markers [21][22]. AGEs activate the RhoA/Rho kinase pathway, recruiting AP-1-mediated transcription of α-SMA. This pathway is active in human peritoneal mesothelial cells as demonstrated by using inhibitors of the RhoA/Rho kinase (Y27632) and curcumin, a compound that has been shown to inhibit AP-1 [23]. The production of reactive oxygen species (ROS) is also involved in MMT [24][25]. IL-17A activates these signaling systems, including RhoA/Rho kinase, ROS production, and the MAPK cascade in different cell types, such as vascular smooth muscle cells [26][27][28][29][30], but data on mesothelial cells are lacking.
A majority of MMT responses converge in Snail expression, which is the principal driver of mesothelial cell junction disruption and loss of basopolarity, leading to a mesenchymal phenotype [31][32]. Among others, Snail expression is regulated by NF-κB, illustrating the key role of this inflammatory transcription factor in MMT. In response to the co-stimulation of TGF-β1 and IL1-β, NF-κB activation is required for E-cadherin and cytokeratin downregulation in mesothelial cells [31]. Inhibition of transforming growth factor-activated kinase-1 (TAK1) blocks the MMT changes caused by activation of NF-κB and Smad3 [33]. Moreover, the p38 MAPK signaling pathway modulates the TAK1-NF-κB pathway [34]. The MAPK kinase (MEK)-extracellular signal-regulated kinase (ERK) signaling pathway can also activate Snail expression. In this case, caveolin deficiency played a role in mesothelial cell transdifferentiation through overactivation of MEK-ERK signaling [35].
IL-17A is known to induce phenotype changes in several cell types. In cultured human tubular-epithelial cells, these changes include loss of the epithelial marker E-cadherin and induction of a myofibroblast-like morphology in a process known as epithelial-to-mesenchymal transition (EMT), associated with proinflammatory and profibrotic factors upregulation [36]. IL-17-induced EMT promoted lung cancer cell migration and invasion via the NF-κB signaling pathway [37]. We have recently described that IL-17A also induced phenotype changes in vascular smooth muscle cells from a contractile to a synthetic cell type, leading to changes in the secretome, including upregulation of pro-inflammatory genes, like MCP-1 and IL-6. However, IL-17A did not increase ECM production in vascular smooth muscle cells [38]. Moreover, other studies in these cells have shown that IL-17A activates some MMT-related pathways, including ROS production, and activation of NF-κB and protein kinases, including RhoA/Rho-kinase and the MAPK cascade [26][27][39][40][30]. Interestingly, in experimental PDF exposure, the IL-17A blockade prevented the induction of MMT markers, such as α-SMA, and peritoneal fibrosis [2][41]. Thus, future studies are needed to assess whether IL-17A can directly induce MMT or can regulate this process in human mesothelial cells.
Numerous preclinical studies have investigated the effect of IL-17A on experimental fibrosis by using different approaches, including deleting the genes of the cytokine or its receptors or blocking cytokine actions by neutralizing anti-IL-17A antibodies, but contradictory results were found. The IL-17A blockade attenuated fibrosis in some experimental models, such as lung [42][43][44], inflammatory skin [45], and intestinal fibrosis [46]. However, data of preclinical kidney damage models showed that fibrosis may be both decreased or increased by IL-17A inhibition/deletion [3][47][48][49]. Our group has demonstrated that systemic administration of IL-17A in mice for 2 weeks increased blood pressure and induced kidney inflammation but had no effect on renal collagen accumulation [50][38]. Consistent with these findings, an IL-17A neutralization treatment did not improve angiotensin II-induced experimental renal or aortic fibrosis [50]. A possible explanation could be a differential response to IL-17A in ECM production by different cell types. In fibroblast cell lines, IL-17A increased ECM synthesis [3], but this was not the case in vascular smooth muscle cells [38]. Several studies have observed that IL-17A responses can be modified in the presence of other cytokines and growth factors, showing either synergistic proinflammatory effects on endothelial cells [51] or an inhibitory effect on profibrotic responses on fibroblasts. In this respect, in human systemic sclerosis skin fibroblasts, IL-17A reduced TGF-β1-induced collagen production and α-SMA expression [52] and downregulated CTGF expression [53].
Additionally, the cell source of IL-17A and physio/pathological context may be relevant. In murine bleomycin-induced pulmonary fibrosis, IL-17A+/γδ+ T cells prevented pulmonary fibrosis, apparently through attenuation of interstitial inflammation and improving epithelial regeneration. In accordance, γδ-deficient mice exhibited increased pulmonary inflammation and ECM deposition [54]. Neutrophils are also a source of IL-17A production. At this point, it is important to highlight the possible contribution of IL-17A to ECM degradation by regulating matrix metalloproteinases (MMPs), mainly produced by neutrophils [49]. In experimental models of renal damage, IL-17 receptor knockout mice presented exacerbated renal fibrosis associated with lower neutrophil but not macrophage infiltration and diminished MMP-2 activity [49]. The authors hypothesized that IL-17 could protect against renal fibrosis by inhibiting the kallikrein-kinin system [55]. Accordingly, systemic IL-17A administration increased renal the kallikrein-1 gene and protein levels, associated with kidney neutrophil infiltration, in the absence of ECM accumulation [50]. Altogether, these data suggest the complex role of IL-17A in the regulation of ECM synthesis/degradation and the importance of understanding the role of IL-17A in fibrosis in each individual disease. Therefore, further studies to clarify this point are needed.
Regarding peritoneum, IL-17A could contribute to peritoneal fibrosis by direct effects on resident fibroblasts. As pointed out before, peritoneal IL-17A administration to mice induced peritoneal fibrosis, characterized by fibronectin accumulation and submesothelial FSP-1 and α-SMA-stained cells [2]. The origin of these activated myofibroblasts was not evaluated in this study, but several sources have been proposed, like resident fibroblasts, phenotype conversion of mesothelial (MMT), or endothelial (EndoEMT) cells to mesenchymal cells, or infiltrating bone marrow-derived cells [56]. Importantly, in two murine models of PDF exposure, treatment with a neutralizing anti-IL-17A antibody inhibited peritoneal fibrosis; decreased the number of α-SMA expressing cells; and diminished the production of profibrotic factors such as TGF-β1, CTGF, and plasminogen activator inhibitor-1 (PAI-1) as well as extracellular matrix components, such as collagens and fibronectin [57][2]. All these processes promoted by IL-17A contribute to PM thickening and to peritoneal fibrosis progression, suggesting a potential key role of IL-17 in PM fibrosis.
Peritonitis, an infection within the peritoneal cavity mainly caused by bacteria, is the most frequent complication of PD [58]. Measures to limit infection risk and to ensure prompt and appropriate investigation and treatment have lowered peritonitis rates and improved outcomes, but peritonitis remains a major determining factor in mortality and in adverse outcomes, including peritoneal inflammation and membrane failure [59]. The immune response to infectious peritonitis is initially characterized by neutrophil recruitment, with subsequent transition to monocyte predominance [60]. These changes are associated with increased intraperitoneal levels of inflammatory cytokines and neutrophil number [61]. Various studies in mice have demonstrated the importance and pleiotropy of IL-17A in the peritoneal inflammatory response during infection. In one such study, abscess formation after infection or surgical injury was preceded by an increase in the number of Th17 cells in the peritoneal cavity, and treatment with neutralizing antibodies against IL-17 prevented the formation of the abscesses [62]. In murine peritonitis, γδ T lymphocytes are the main source of IL-17A [63][64]. Elevated intraperitoneal IL-17A levels following caecal ligation and puncture are found in mice, and intraperitoneal IL-17A blockade decreased proinflammatory cytokine production in the peritoneal cavity and caused subsequent lung injury, thus improving mouse survival [64]. In patients, the cytokine profile evident during an episode of peritonitis may predict the outcome. For example, high levels of IL-12 and IL-18 may be evident during the early phase of peritonitis and may correlate with a predominant type 1 immune response and recovery [65]. IL-17A is typically present at very low levels in peritoneal dialysis fluid (PDE) from uninfected patients and may increase many-fold during acute peritonitis [66]. High levels of intraperitoneal IL-17A have been correlated with favorable outcomes in PD-associated peritonitis [67]. This may suggest a protective role of IL-17A in early immune response in the peritoneal host defense but may also reflect the better outcomes seen following gram-positive bacterial infections, the class of organism where high levels of intraperitoneal IL-17A are typically seen [68].
Macrophages play a key role in the correct function of the PM, as they modulate peritoneal inflammation and fibrosis [69][70][71]. Classically, macrophages were divided into 2 subtypes, M1 or classically activated and M2 or alternatively activated, based on cytokine expression profiles and surface markers. However, recent data suggest the existence of many mixed phenotypes depending on pathological conditions [72]. M1 macrophages produce pro-inflammatory factors such as IL-1β, TNF-α, IL-6, IL-23, IL-18, IL-12, and CXCL10; activate inducible nitric oxide synthase (iNOS); produce ROS; and develop cytotoxic properties [73][74][75]. M2 macrophages express indoleamine 2,3-dioxygenase, arginase I, and mannose receptor and release cytokines, like decoy IL-1RII, CCL-17, CCL-18, CCL-22, the anti-inflammatory cytokine IL-10, and profibrotic growth factors such as TGF-β1 or VEGF [76][77]. Currently, there is no clear correspondence between these human subtypes and murine macrophages due to the existence of overlapping phenotypes and different surface marker expression in different species, thus complicating the extrapolation of preclinical studies to human diseases [78]. Moreover, recent studies have increased the complexity of these classifications [72].
In PD patients, alterations of macrophage heterogeneity, characterized by different maturation and activation states, have been associated with different PD outcomes [79]. Thus, an increased proportion of the CD16−CD206− macrophages subtype was founded in gram-negative peritonitis and failed peritonitis treatment, whereas an increased proportion of CD16+CD206− macrophages subtype was observed in “new-starter” patients with catheter failure and stable patients with a history of recurrent peritonitis episodes [79].
Peritoneal macrophages isolated from PDE of patients under continuous ambulatory PD (CAPD) during peritonitis episodes showed higher production of the proinflammatory cytokines IL-1β and TNF-α than infection-free macrophages [80]. Later studies in PDE from PD patients showed that peritoneal M2 macrophages (CD206+ and CD163+) participate in peritoneal fibrosis by favoring fibroblast overgrowth and increased CCL-18 production [81]. CCL-18 is a cytokine mainly produced by M2 macrophages associated with fibrosis/tissue repair and is increased in PDE of patients with peritonitis episodes [82][83]. Additionally, in a model of encapsulated peritoneal sclerosis, it was noted that inflammatory M2 macrophages switch to the profibrotic phenotype and activate peritoneal fibroblasts through CCL-17 after sodium hypochlorite-induced injury [84]. In a model of macrophage depletion in PDF-exposed mice, transfusion of macrophages of distinct phenotypes showed a pathogenic role for M1 macrophages. M1 macrophages increased peritoneal fibrosis and disturbed peritoneal ultrafiltration more than M2 macrophages [85]. Another study of experimental PDF exposure described an increase of peritoneal thickness; fibrotic markers including collagen type I; fibronectin; and the M2 macrophage subtype markers CD206, TGF-β, Ym-1, and Arg-1. These effects were recovered by treatment with a liposome-encapsulated clodronate (LC, a specific scavenger of macrophages) [86]. A recent study demonstrated that dialyzed patients have a significantly lower content of Omega-3 fatty acids, such as n-3 Polyunsaturated fatty acid (PUFA), and this situation contributes to a high cardiovascular risk in CKD patients [87]. A study in an experimental model of PD in rats showed that the treatment with n-3 PUFA reduced peritoneal fibrosis through inhibition of activated fibroblasts and M2 macrophages [88].
As commented before, PDF exposure models showed that IL-17A neutralization decreased submesothelial macrophage infiltration, but macrophage phenotypes and cytokine profiles were not characterized [2][41]. Another study observed that, in uremic mice, exposure to standard PDF (lactate-buffered solution) increased M1 macrophages and CD4+/IL-17+ cells in PDE [89]. In this regard, IL-17A modulates monocyte/macrophage functions such as monocyte migration, promotion of cytokine production [90][91], and macrophage phenotype modulation. In cultured macrophages derived from human THP-1 monocytes, stimulation with IL-17A increased the gene expressions of VEGF, TGF-β1, and IL-10 and upregulated M2 macrophage markers, such as CD206, CD163, Arginase I, Ym1 (also known as chitinase 3-like 3), and Fizz1 (also known as resistin-like beta) [92]. In these cells, IL-17A-induced M2 polarization was mediated through NF-κB signaling [92]. Preclinical studies confirmed the potential role of IL-17A on M1/M2 macrophage differentiation. In lung cancer cells, increased levels of IL-17A and PGE2 were involved in the development of an M2-macrophage-dominant tumor microenvironment [93]. In human and murine jawbone osteonecrosis, IL-17A mediated the M1 polarization of macrophages, and serum IL-17A levels correlated with the M1/M2 macrophage ratio at the lesion foci [94]. In other diseases, such as endometriosis, IL-17A induced pathological macrophage polarization into the M2 phenotype [95]. In contrast, IL-17A-deficient mice with severe colitis presented milder intestinal inflammation and decreased M2-like macrophages, suggesting a potential beneficial effect of IL-17A in colitis [96]. In a mouse model of lipopolysaccharide (LPS)-induced peritonitis, macrophage polarization was linked to the development and progression of infection through the JAK/STAT signaling pathway [97]. Mice with E. coli peritonitis showed an increased IL-17A expression in immune cells including CD11b+ and CD11b− neutrophils, macrophages, and CD3+ T cells [98]. In patients with cirrhosis and peritonitis, serum levels of the M2 macrophage marker CD206 were increased and associated with mortality risk [99]. In conclusion, these sometimes-controversial results require a more in-depth analysis of the specific cellular and molecular mechanisms that drive the deleterious or beneficial effect of IL-17A in macrophage polarization associated with specific pathological environments and, finally, elucidate the role of IL-17A in determining macrophage phenotype in stable PD patients or during peritonitis episodes.
One of the specific changes observed after chronic peritoneal exposure to PDF is an increased number of capillaries (angiogenesis), which is driven by VEGF and linked to an increased PM permeability [100][101][102]. In this context, mesothelial cells acquire the capacity to synthesize pro-inflammatory and pro-angiogenic molecules, such as VEGF [100], turning them into the main local source of VEGF during PD [11]. In cultured omentum-derived mesothelial cells, stimulation with TGFβ-1 and IL-1β to induce MMT resulted in the downregulation of the two most important VEGF receptors, VEGFR1 and VEGFR2, whilst the co-receptor neuropilin-1 (Nrp1) was increased. Therefore, during in vitro MMT, the VEGF/Nrp1 interaction drives mesothelial cell behavior [103].
The potential role of IL-17A in angiogenesis induction was evaluated in proliferative disorders. The number of infiltrating IL-17A-secreting cells directly correlated with microvessel density in tumors [104][105][106]. In this regard, in human colorectal carcinoma, IL-17A has been identified as an indicator of poor prognosis [106]. Accordingly, IL-17A and VEGF serum levels in patients with lung adenocarcinoma were positively correlated [107], and in tumoral cell lines, IL-17A induced VEGFA expression [106]. IL-17A-induced VEGF expression seems to be mediated by STAT3 [108][109] or STAT1 [107], but further clarification is needed concerning the molecular pathways involved and their contribution to angiogenesis. Additionally, IL-17A can also indirectly cause angiogenesis and neovascularization by stimulating the production of additional proangiogenic factors, including chemokines such as CXCL1, CXCL5, CXCL6, and CXCL8 [57][110][111]. These chemokines activate the CXCR2 receptor in endothelial cells to promote migration and proliferation [112][113]. In this regard, CXCL1 can activate VEGF signaling in gastric tumor cells [114] and CXCL8 activation of CXCR2 increased VEGF mRNA expression in cultured endothelial cells [115], suggesting a relation between proangiogenic chemokines, produced in response to IL-17A, and VEGF expression. Further studies are needed to explore the mechanisms by which IL-17A promotes VEGF expression, specifically in peritoneum exposed to PDF as well as its contribution to angiogenesis in this context (Figure 2).





